Окостеніння черепних швів. Зростання мозкового черепа
. - С. 18-23.
УДК 340.84-053:616.714.14-007.11
Науково-дослідний інститут судової медицини (дир. – проф. В.І. Прозоровський) Міністерства охорони здоров'я СРСР, Москва
Передчасне заростання черепних швів у судово-медичному аспекті. 3вягін В.М. Суд.-мед. експерт., 1967 № 3, с. 18
Досліджено колекції черепів російського (864) та бурятського (180) населення. В цілому чоловіки більш схильні до краніостенозу. Впливу етнічної власності не виявлено. Отримані результати доводять необхідність диференційованого підходу до «шовного заростання» щодо віку по черепу.
PREMATURE OBLITERATION OF CRANIAL SUTURES: THE MEDICO-LEGAL ASPECT
На рисах різних статей craniostenosis були вивчені craniological collections of modern Russian (864) and Buryat (180) populations. Як і всі, люди можуть більше бути досліджені до патології. Результати охоплюють show a needity of different viewpoint on “suture obliteration” in age estim ation by human skulls.
Передчасне заростання черепних швів у судово-медичному аспекті
бібліографічний опис:
Передчасне заростання черепних швів у судово-медичному аспекті / Звягін В.М. // Судово-медична експертиза. - М., 1976. - №3. - С. 18-23.
html код:
/ Звягін В.М. // Судово-медична експертиза. - М., 1976. - №3. - С. 18-23.
код для вставки на форум:
Передчасне заростання черепних швів у судово-медичному аспекті / Звягін В.М. // Судово-медична експертиза. - М., 1976. - №3. - С. 18-23.
wiki:
/ Звягін В.М. // Судово-медична експертиза. - М., 1976. - №3. - С. 18-23.
Визначення віку дорослої людини по черепу проводять за станом зубного апарату та швів. У деяких випадках показання критеріїв бувають суперечливими. Тоді перевага одному з них певною мірою буває інтуїтивною.
Мета цієї статті - звернути увагу до передчасне заростання швів, т. е. ті випадки, коли користуватися критерієм «шовного заростання» не можна.
Це питання в судово-медичній літературі висвітлено недостатньо. Література з рентгенології та хірургії (Bolk, 1915; В.А. Дьяченко, 1954; В.А. Козирєв, 1962 та ін) не заповнює цю прогалину.
Досліджували краніологічні серії російського та бурятського населення, що відноситься до європеоїдної та монголоїдної рас. Російські серії представлені черепами колекції Таренецького у Військово-медичній академії (чоловіків 140, жінок 58), Інституту судової медицини (чоловіків 42, жінок 28) та інших судово-медичних установ Москви (чоловіків 24, жінок 34), матеріалами автора з території Кіровської чоловіків 194, жінок 108) та Московської (чоловіків 93, жінок 143) областей. Паспортні дані відомі. Бурятське населення представлене матеріалами Музею антропології та етнографії Академії наук СРСР (чоловіків 98, жінок 82). Серії не паспортизовані, тому статеву приналежність та вік визначали додатково.
Специфічною та найбільш стійкою ознакою є передчасне окостеніння одного або декількох швів. Воно виражається повним зникненням шовної лінії та відсутністю диплоїчного зубця (у нормі шов зовнішньої платівки зберігається все життя).
Менш постійною ознакою є деформація мозкового та лицьового скелета. Можуть спостерігатися різні її форми.
До іншої групи відносять морфологічні зміни, що виникають в результаті невідповідності зростаючого мозку об'єму черепа:
1) поглиблення судинних борозен і синусів, зменшення кількості диплоїчних вен та їх розширення, розвиток множинних венозних випускників; тканини остеонними конструкціями.
Таблиця 1
Частота народження (в %) деяких різновидів краніостенозу
Передчасне заростання черепних швів (табл. 1) трапляється досить часто, переважно у чоловіків. Зв'язку його з етнічною приналежністю ми не відзначили, як і Bolk (1915), який отримав близькі частоти у голландських дітей.
Процес, як правило, виникає в ранньому дитинстві і супроводжується деформацією черепа: у російських і бурятських чоловіків чин-11,49 і 11,22%, у жінок - відповідно 8,89 і 8,52%.
Прискорене заростання швів після 7-8 років, яке не супроводжується деформацією, зустрічається у чоловіків і жінок однаково часто (2,69-4,43%).
Зустрічаність асиметричних деформацій дуже варіабельна. По відношенню до всіх деформацій вони мають більшу питому вагу у жінок - росіян 48,48%, буряток 57,14%, у чоловіків відповідно 42,63% і 36,36%. У чоловіків частота стенозування лівосторонніх швів вища (у російських 3,97%, у бурятів 3,06%), ніж правосторонніх (1,81% і 1,02%). У жінок картина зворотна (у російських жінок 1,35%, у чоловіків 2,96%, у буряток 1,22% У бурят 3,66%).
1. Передчасна облітерація поперечних швів
Вінцевий краніостеноз(Сфеноцефалія). Череп високий, короткий та відносно широкий. Темні бугри виражені чітко, лобові - помірковано. Лоб досить прямий і плоский, вибухає область великого джерельця. Потилиця лок укорочена, висота його вигину мала. Ока високі. Склепіння брихікранне. Черепні ями заглиблені. У російських деформація виникає з частотою 0,4% у хлопчиків і 0,54% у дівчаток (по Bolk - 0,3%) 1 .
Потиличний краніостеноз (товсті черепи). Поздовжній діаметр різко зменшено, поперечні та висотні розміри мозкового черепа збільшені або без змін. Лоб скошений. Темні кістки сильно вигнуті над областю ламбди. Луска потиличної кістки значно сплощена. Задня яма черепна дрібна. Зустрічаємо краніостенозу у російських чоловіків 0,6%, у жінок 0,27%; причому жіночі черепи та 0,4% чоловічих не мають деформацій.
Вінцево-потиличний краніостеноз (лептоцефалія, коротка голова). Поздовжні розміри черепа укорочені, поперечні дещо збільшені. Лобові горби не розвинені. Кривизна лобової та потиличної кісток незначна. Бічні поверхні черепа вибухають. Черепні ями, за винятком задньої, дрібні. Частота краніостенозу, мабуть, менше 1:1000.
2. Передчасна облітерація поздовжньо розташованих швів
Лобний краніостеноз (тригоноцефалія, клиноцефалія, клиноподібна голова, трикутний череп). Ізольоване закриття лобового (метопічного) шва раніше 2-4 місяців життя веде до освіченого загостреного допереду черепа. Лобова кістка різко відхилена назад, з гострим гребенем по сагітальній лінії. Темні бугри різко виступають. Задні відділи черепа компенсаторно збільшені та опущені, що пов'язано із розширенням задньої черепної ями. Цей вид деформації спостерігали лише у російських чоловіків – у 0,81 %.
Стрілоподібний краніостеноз (скафоцефалія, човноподібний череп, кілеголовість). Частота народження по Bolk 2,5%. Мозковий череп відносно низький, вузький та дуже довгий. Серединний контур склепіння з різко опуклим чолом і потилицею. Висота вигину темряви незначна. Зведення еліпсоїдний, звужений від птеріону. Передня та середня ями подовжені та поглиблені. Деформація однаково часто зустрічається у російських чоловіків та жінок (0,81%). Поступове і пізнє закриття стрілоподібного шва не змінює форми черепа і відзначається у 2,42% чоловік чин і у 1,35% жінок.
Лускатий краніостеноз. Зарощення правого та лівого лускатого швів формує помірно долихокранні черепи з широким опуклим лобом та ортогнантним обличчям. Нерідко вибухає луска скроневих кісток. У російських краніостеноз спостерігали у 0,6% чоловіків та 0,27% жінок. За даними Bolk, частота дорівнює 0,15%.
3. Поєднана передчасна облітерація поперечно- і поздовжньо розташованих швів
Вінцево-стрілоподібний краніостеноз (акроцефалія, турикцефалія, баштовий череп). Череп дуже високий, циліндричний. Лоб сплощений, крутий чи нависаючий. Область великого тім'ячка у вигляді кісткового бугра. Обличчя широке та довге. Склепіння з клиноподібною потилицею. Ями заглиблені. При ранньому заростанні швів – у 0,4% чоловіків та 0,27% жінок – деформація виражена значно. Пізніше закриття швів (0,6% у чоловіків та 0,27% у жінок) не змінює черепа.
Стріловидно-птеріонний, стрілоподібно-астеріонний краніостеноз. Передчасне закриття стрілоподібного шва з симетричною облітерацією птеріонних (сідлоподібний череп) або астеріонних швів (горбоподібні черепи) сприяє утворенню довгохранної або мезокранної конфігурації. Сідлоподібна деформація виражається різким звуженням кісток позаду вінцевого шва. Нерідко відбувається горбоподібне випинання верхівкової області та тім'яних пагорбів. Лобова кістка значно сплощена. Сагітальний контур заднього відділу черепа вигнутий слабо. Підстава страждає трохи. Частота деформації у чоловіків – росіян 1,81%, бурятів 2,04%, у жінок – російських 2,16%, буряток 2,44%. Загальний краніостеноз (оксицефалія, гостра голова, «цукрова голова»). При поєднаному зарощенні вінцевого, стрілоподібного та потиличного швів череп відносно високий при зменшених поперечному і особливо поздовжньому діаметрах, догори помітно звужений. Луска скроневих кісток витончена, сильно вибухає. Вилицьові дуги зазвичай деформовані. Високі орбіти, розділені широким перенесенням (гіпертелоризм), ємність зменшена. Різко виражений прогнатизм. Верхні та нижні різці не стикаються через зміщення взад укороченого альвеолярного відростка нижньої щелепи (опістодонтія). Основа черепа втиснута. Тіло потиличної кістки вигнуте опуклістю вниз. Оксицефалічну деформацію спостерігали лише у російських чоловіків у 0,4%, з них половину склали випадки поєднання із зарощеними лускатими швами. Пізніше закриття всіх швів спостерігали у 0,4% чоловіків та 0,54% жінок.
Таблиця 2
Характеристика основних видів краніостенозу у російського населення за 7-бальною шкалою
4. Асиметричні деформації
Передчасне право-або лівостороннє окостеніння вінцевого та потиличного швів, швів бічної поверхні – лускатих, астеріонних, птеріонних – веде до появи асиметричних форм черепа (плагіоцефалія, косі черепа). Процес закриття рідко залишається ізольованим, він захоплює граничні шви. При плагіоцефалії відбуваються поворот площини обличчя у бік зарослого шва та компенсаторне сплощення протилежної тім'яно-потиличної області. При вираженій деформації відбувається дугоподібне викривлення стрілоподібного шва та сагітальної борозни опуклістю від зарослого шва. Асиметричні деформації бувають при одночасному закритті зазначених груп швів зі стрілоподібним та в різних поєднаннях один з одним. У ряді випадків відзначається ізольоване порушення розташування стрілоподібного шва - косе, зигзагоподібне і т.д.
Надалі для усунення суб'єктивізму ми спробували використати груповий профіль Моллісона з розбивкою відхилень розмірів від базису на 7 категорій. Базисом обрали дані В.П. Алексєєва (1969) за російським населенням, за ознаками 23а, 24, 25, 34, 27, 28, 30 і 31 (номери за Мартіном) - дані М.С. Великановій (1974). Мерою відхилення взято середній стандарт (ст) для сучасного людства (В.П. Алексєєв, Г.Ф. Дебець, 1964). Межі категорій у частках сигми наведено у табл. 2.
7-бальна характеристика основних видів краніостенозу за комплексом 16 ознак відображає морфологічний опис, вільна від елементів довільності та полегшує взаємне порівняння. Зіставлення теоретично передбаченої (за інтегралом ймовірностей) і дійсної частоти попадань розмірів у певну категорію статистичних відмінностей не показало (х 2 = 9,08 при х 2 пороговому = 12,59). Отже, деформація черепа при краніостенозі в цілому відбувається в рамках фізіологічного розкиду за окремими ознаками.
Діагноз краніостенозу, що виник у ранньому дитинстві, нескладний завдяки наявності всього комплексу ознак. Після 7-8 років, коли мозок досягає 95% свого обсягу, діагностування захворювання ускладнене, оскільки зміна величини черепа, його деформація та ознаки підвищеного внутрішньочерепного тиску можуть бути відсутніми. У подібних випадках особливу диференціальну значущість набувають ознак першої групи, які необхідно ретельно дослідити рентгенографічним та мікроскопічним методами. Використання для діагностики віку критерію «шовного заростання» у разі було б грубої помилкою.
1 Зустрічаність окремих різновидів краніостенозу у бурятів через невеликий склад вибірки дана лише за досить великої їх частоті.
Березень, 2007
А.В. Лопатін, С.А. Ясонов, відділення щелепно-лицьової хірургії, ГУ «Російська дитяча клінічна лікарня»
Практично будь-який лікар курсу загальної анатомії пам'ятає про існування специфічних форм черепа, таких, як скафоцефалічний (витягнутий у передньо-задньому напрямку) та брахіцефалічний (збільшений завширшки). Але рідко хто згадує у тому, що незвичайна форма черепа в дитини у часто є ознакою передчасного зарощення черепних швів.
Звичайно, всі лікарі знайомі з терміном краніостеноз - передчасне зарощення швів черепа, що призводить до неспецифічного пошкодження головного мозку через недостатнє розширення порожнини черепа в період найактивнішого росту мозку. Коли виникає питання про те, як лікують краніостеноз, деякі згадують про можливість хірургічного висічення передчасно зарослих швів і лише одиниці знають про існування методу двошкутної краніотомії.
Тим часом, за міжнародною статистикою, передчасне закриття одного із швів черепа (ізольований краніосиностоз) виникає приблизно у одного із 1000 дітей. Цікаво відзначити, що така ж частота характерна і у дітей, які мають ущелину губи. При цьому ні в кого не викликає труднощів діагностика ущелин губи, тому що таких пацієнтів, безперечно, бачив кожен лікар. Тоді як майже ніхто з лікарів загальної практики не може пригадати, чи бачив він колись дитину з передчасним зарощенням швів черепа.
Ходіння по муках
Краніосиностоз - передчасне зарощення одного або декількох швів черепа, що призводить до формування характерної деформації голови. Таким чином, вже в пологовому будинку дитина з підозрою на краніосиностоз може бути виділена із загальної маси новонароджених та спрямована на дообстеження. Насправді, на жаль, цьому етапі все деформації черепа, виявлені в дітей віком, розцінюються лікарями як особливості післяпологової зміни голови і не приділяється належної уваги. У період новонародженості формі черепа також не надається великого значення. Звичайна відповідь педіатра на стурбованість батьків: «... Нічого страшного, добре додає у вазі, пішла жовтяниця, а голова така від того, що лежить на боці. Ось почне ходити і все зникне».
Психомоторний розвиток дітей відбувається з відставанням, деформації черепа спонтанно не зникають, деякі деформації стають менш помітними, ховаючись під волоссям, інші помилково розцінюються лікарями як інші захворювання, а треті відступають на другий план за наявності очевидніших порушень функції органів і систем. Найчастіше малюків із краніосиностозами консультують генетики, і нерідко правильно встановлюється група захворювань і навіть передбачається безпосередній генетичний синдром. Незважаючи на це, одиниці таких пацієнтів надходять до спеціалізованих клінік для проведення лікування. Переважна більшість дітей, які не отримали лікування, мають знижений інтелект та стають інвалідами. Через незвичайну форму черепа порушуються пропорції особи, і до періоду статевого дозрівання у дітей частіше, ніж в інших, виникають труднощі у соціальному спілкуванні і навіть можливі суїцидальні спроби.
Батьки поступово перестають звертати увагу на легкі деформації черепа, а за наявності вираженої у дитини деформації обличчя дитячі хірурги пояснюють їм, що виправлення косметичних дефектів проводиться лише у 16 років.
Виходячи зі сказаного, можна зробити висновок, що в нашій країні практично повністю відсутня кваліфікована допомога дітям з передчасним зарощенням одного або декількох швів черепа. При цьому в усьому світі протягом останніх сорока років лікуванню дітей із вродженими деформаціями черепа приділяється дуже велика увага, а розроблені методики оперативного лікування дозволяють усунути компресію мозку та значно покращити зовнішній вигляд дітей із краніосиностозами вже у трьох-шестимісячному віці. Основною причиною такого пропуску є відсутність доступної інформації щодо особливостей діагностики та лікування вроджених деформацій черепа у дітей. Для того, щоб трохи виправити існуюче положення, пропонуємо розглянути найзагальніші питання діагностики та лікування краніосиностозів.
Діагностика
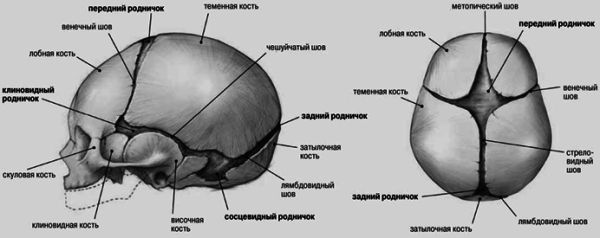
Основними швами склепіння черепа є сагітальний, коронарний, лямбдоподібний та метопічний (рис. 1). При передчасному зарощенні кісткового шва відбувається компенсаторне зростання кісток перпендикулярно його осі (закон Вірхова). Внаслідок цього з'являється характерна деформація. Опишемо форми краніосиностозів, що найчастіше зустрічаються.
Сагітальний краніосиностоз
Передчасне зарощення сагіттального шва призводить до збільшення передньо-заднього розміру черепа з нависаючими лобовою та потиличною областями та зменшення його ширини з формуванням вузького овального обличчя (рис. 2). Такий вид деформації називають скафоцефалією, або човноподібним черепом. Це найчастіше захворювання серед загальної кількості ізольованих синостозів (50-60%). Характерна форма черепа видно вже від народження. При огляді голови зверху помітне втягнення тім'яних областей, це дає відчуття циркулярної перетяжки склепіння черепа на рівні або трохи позаду вушних раковин. Чітко визначається велике тім'ячко, причому його розміри не відрізняються від норми. Характерним вважається наявність кісткового гребеня, що пальпується у проекції сагіттального шва.

: а - дитина до і б - після усунення скафоцефалії
Метопічний краніосиностоз
Найрідкіснішим представником групи ізольованих краніосиностозів є метопічний краніосиностоз, або тригоноцефалія, що становить 5-10% від загальної їх кількості. Незважаючи на це, це захворювання, мабуть, найчастіше розпізнається як уроджена деформація черепа через характерну клінічну картину.
При ранньому замиканні метопічного шва відбувається формування трикутної деформації чола з утворенням кісткового кіля, що йде від надперенесення до великого джерельця. При погляді на такий череп зверху видно чітку трикутну деформацію з вершиною в області надперенесення. При цьому верхні та латеральні краї орбіт зміщуються дозаду, що дає відчуття розвороту площини орбіт назовні та зменшення міжорбітальної відстані (гіпотелоризм). Деформація чола настільки незвичайна, що діти з тригоноцефалією часто обстежуються у генетиків і спостерігаються як носії спадкових синдромів, які супроводжуються зниженням інтелекту. Справді, тригоноцефалія сприймається як невід'ємна частина таких синдромів, як Opitz, Oro-facio-digital syndrom, та інших. Правильно і те, що багато синдромальних захворювань призводять до затримки інтелектуального розвитку, але частота їх настільки низька, а клінічна картина настільки характерна, що не варто всіх дітей, які мають лише метопічний синостоз, зараховувати до групи ризику розвитку розумової неповноцінності.
Односторонній коронарний краніосиностоз
Коронарний шов розташований перпендикулярно до серединної осі черепа і складається з двох рівноцінних половин. Так що при передчасному зарощенні однієї з його половин формується типова асиметрична деформація, що має назву плагіоцефалія. Вид дитини з плагіоцефалією характеризується ущільненням верхньоорбітального краю орбіти та лобової кістки на стороні поразки з компенсаторним нависанням протилежної половини чола (дитина ніби хмуриться однією стороною обличчя). З віком виразніше починає проявлятися іпсилатеральне сплощення вилицьової області і викривлення носа в той же бік. У шкільному віці приєднується деформація прикусу, пов'язана зі збільшенням висоти верхньої щелепи і як наслідок – зміщенням нижньої щелепи на стороні шва, що передчасно закрився. У важких випадках є навіть компенсаторне вибухання потиличної з боку синостозу. Порушення з боку органу зору представлені найчастіше односторонньою косоокістю. Плагіоцефалія найчастіше розцінюється як особливості післяпологової зміни голови. Але, на відміну від останньої, вона не зникає в перші тижні життя, а, навпаки, з віком прогресує.
Отже, величезну роль правильної постановці діагнозу грає саме форма черепа.
З інструментальних методів діагностики найкращим є проведення комп'ютерної томографії з тривимірним ремоделювання зображення кісток склепіння черепа та обличчя. Це обстеження допомагає виявити супутню патологію головного мозку, підтвердити наявність синостозу у разі ізольованого пошкодження та встановити всі зацікавлені шви у разі полісиностозу.
Лікування
Найактивнішим періодом зростання мозку вважається вік до двох років. Таким чином, з функціональної точки зору запобігти краніостенозу можна раннім хірургічним лікуванням. Оптимальним віком щодо операції з приводу краниосиностоза вважатимуться період із 3 до 9 місяців. Перевагами лікування у цьому віці можна вважати:
- легкість маніпулювання з тонкими та м'якими кістками черепа;
- полегшення остаточного ремоделювання форми черепа швидко зростаючим мозком;
- більш повне та швидке загоєння залишкових кісткових дефектів.
Якщо лікування виконується після п'яти років, то сумнівно, що воно призведе до значного поліпшення функції головного мозку. Більшою мірою операція буде спрямована на усунення деформації голови.
Основною особливістю сучасного хірургічного лікування є не тільки збільшення об'єму черепа, а й виправлення його форми та поєднаної деформації обличчя під час однієї операції.
Ще раз звертаємо увагу читачів на те, що краще перестрахуватися та направити дитину з деформацією черепа до фахівця, ніж пропустити патологію. Відділення щелепно-лицьової хірургії Російської дитячої клінічної лікарні накопичило великий досвід у діагностиці та лікуванні цих станів у дітей. Маленькі пацієнти вступають у відділення з усіх регіонів Росії. Для того, щоб потрапити на прийом до фахівця, необхідно звернутися до консультативного відділення лікарні. Бажано мати напрямок регіонального управління охорони здоров'я. Якщо дитині дійсно потрібна хірургічна допомога, намічається план обстеження і видається путівка на госпіталізацію, в якій зазначена дата надходження до стаціонару та перелік усіх необхідних документів. Термін перебування у клініці становить у середньому 34 тижні. Діти можуть перебувати у відділенні з одним із батьків. Усі діти після проведеного лікування до 18-річного віку перебувають під наглядом лікарів відділення 1 .
Узагальнюючи вищевикладене, ще раз зазначимо: у нашій країні є великий пласт хворих, які через низьку поінформованість медиків про сучасні можливості діагностики та лікування краніосиностозів не отримують адекватної допомоги. Тим часом діагностика таких станів є досить простою і можлива вже на ранніх етапах. Своєчасна кваліфікована допомога дітям із краніосиностозами дозволяє вже у перші місяці життя усунути не лише функціональний дефіцит, а й виправити супутню косметичну деформацію.
Андрій В'ячеславович Лопатін, зав. відділенням щелепно-лицьової хірургії ДУ «Російська дитяча клінічна лікарня» Росздрава, професор, д-р мед. наук
Сергій Олександрович Ясонов, лікар відділення щелепно-лицьової хірургії ГУ «Російська дитяча клінічна лікарня»
1 Більш детальну інформацію можна отримати на сайті: www.cfsmed.ru
Краніосиностоз характеризується передчасним зрощенням одного або більше черепних швів, що часто призводить до ненормальної форми голови. Це може бути результатом первинного неправильного окостеніння (первинний краніосиностоз) або, що найчастіше зустрічається, порушення росту головного мозку (вторинний краніосиностоз).
Захворювання найчастіше виникає внутрішньоутробно або в дуже ранньому віці. Воно піддається винятково хірургічного лікування, хоча позитивний результат можливий не завжди.
Класифікація краніосиностозу та причини його розвитку
Нормальна осифікація склепіння черепа починається в центральній області кожної кістки черепної коробки та проходить назовні до черепних швів. Що вказує на норму?
- Коли вінцевий шов відокремлює дві лобові кістки від тім'яних кісток.
- Метопічний шов відокремлює лобові кістки.
- Сагітальний шов відокремлює дві тім'яні кістки.
- Лямбдоподібний шов відокремлює потиличну кістку від двох тім'яних кісток.
Основним фактором, який стримує несвоєчасне зрощення кісток черепа, вважається зростання мозку. Варто підкреслити, що нормальне зростання кожної черепної кістки відбувається перпендикулярно кожному шву.
- Простий краніосиностоз - це термін, який використовується в ситуаціях, коли тільки один шов зрощується передчасно.
- Термін комплексний, або сполучний краніосиностоз, використовується для опису передчасного зрощення кількох швів.
- Коли діти, що показують симптоми краніосиностозу, також страждають на інші потворності тіла, це називається синдромальним краніосиностозом.
Первинний краніосиностоз
При передчасному зрощенні одного або більше швів зростання черепа може бути обмежене перпендикулярними швами. Якщо кілька швів зрощуються в той час, коли мозок все ще змінюється у розмірах, внутрішньочерепний тиск може збільшитись. І це часто закінчується рядом найскладнішої симптоматики, аж до смерті.
Різновиди первинного краніосиностозу (передчасного зрощення)
- Скафоцефалія – стрілоподібний шов.
- Передня плагіоцефалія – перший вінцевий шов.
- Брахіцефалія – двосторонній вінцевий шов.
- Задня плагіоцефалія – раннє закриття одного ламбдоподібного шва.
- Тригоноцефалія – передчасне зрощення метопічного шва.
Вторинний краніосиностоз
Частіше, ніж при первинному типі, цей різновид патології може призвести до раннього зрощення швів через первинну недостатність росту мозку. Оскільки зростання мозку контролює відстань кісткових пластинок одна від одної, розлад його зростання є основною причиною передчасного зрощування всіх швів.
При цьому різновиді патології внутрішньочерепний тиск, як правило, нормальний, і тут рідко буває потреба в хірургії. Як правило, відсутність зростання мозку призводить до мікроцефалії. Передчасне закриття шва, що не створює загрози росту мозку, також не потребує хірургічного втручання.
Внутрішньоматкові просторові обмеження можуть відігравати певну роль у передчасному зрощенні швів черепа плода. Це було продемонстровано у спостереженнях при корональному краніосиностозі. Інші вторинні причини включають системні розлади, що впливають на кістковий метаболізм, такі як рахіт і гіперкальціємія.
Причини та наслідки раннього краніосиностозу
Декілька теорій було запропоновано для етіології первинного краніосиностозу. Але найбільшого поширення набув варіант з етіологією первинного дефекту в мезенхімальних шарах кісток черепа.
Вторинний краніосиностоз, як правило, розвивається поряд із системними розладами.
- Це – ендокринні порушення (гіпертиреоз, гіпофосфатемія, дефіцит вітаміну D, ниркова остеодистрофія, гіперкальціємія та рахіт).
- Гематологічні захворювання, які викликають гіперплазію кісткового мозку, наприклад серповидно-клітинна хвороба, таласемія.
- Низькі темпи зростання головного мозку, у тому числі мікроцефалія та її основні причини, наприклад, гідроцефалія.
Причини синдромального краніосиностозу полягають у генетичних мутаціях, відповідальних за рецептори факторів зростання фібробластів другого та третього класу.
Інші важливі фактори, які варто враховувати щодо етіології захворювання
- p align="justify"> Диференціація плагіоцефалії, що часто є результатом позиційного зрощення (який не вимагає операції і досить часто зустрічається) від зрощення лямбдоподібного шва, є надзвичайно важливим аспектом.
- Наявність кількох зрощень наводить на думку черепно-лицьового синдрому, який часто потребує діагностичної експертизи у педіатричній генетиці.
Симптоми краніосиностозу та методи діагностики
Краніосиностоз у всіх випадках характеризується неправильною формою черепа, яка у дитини визначається за типом краніосиностозу.
Основні ознаки
- Жорсткий кістковий хребет, що добре пальпується по ходу патологічного шва.
- М'яке місце (джерело) зникає, голова дитини змінює форму, чутливість у цих областях, як правило, змінена.
- Голова дитини не росте пропорційно до решти тіла.
- Підвищений внутрішньочерепний тиск.
У деяких випадках краніосиностоз не може бути помітним протягом декількох місяців після народження.
Підвищений внутрішньочерепний тиск є частою ознакою всіх типів краніосиностозів, за винятком деяких вторинних патологій. Коли тільки один шов зрощується передчасно, підвищений внутрішньочерепний тиск трапляється менше ніж у 15% дітей. Тим не менш, при синдромальному краніосиностозі, де беруть участь кілька швів, підвищення тиску може спостерігатися у 60% випадків.
Якщо дитина страждає на легку форму краніосиностозу, хвороба не може бути помічена, поки пацієнти не починають відчувати проблеми через збільшення внутрішньочерепного тиску. Це зазвичай відбувається у віці від чотирьох до восьми років.
Симптоми підвищеного внутрішньочерепного тиску
- Починаються зі стійкого головного болю, як правило, що погіршуються вранці і в нічний час.
- Проблем із зором - двоїнням, помутнінням зору або порушенням кольорового зору.
- Нез'ясовного зниження розумових здібностей дитини.
Якщо дитина скаржиться на будь-який із перерахованих вище симптомів, слід звернутися до педіатра якнайшвидше. У більшості випадків ці симптоми не будуть викликані підвищеним внутрішньочерепним тиском, але їх необхідно обов'язково вивчити.
За відсутності лікування інші симптоми підвищеного внутрішньочерепного тиску можуть включати:
- блювання;
- дратівливість;
- млявість та відсутність реакції;
- опухлі очі або труднощі в спостереженні за об'єктом, що рухається.
- порушення слуху;
- утруднене дихання.
При уважному розгляді черепа зрозуміло, що його форма який завжди підтверджує діагноз краниосиностоза. У таких випадках застосовують низку методів візуального дослідження, наприклад, рентгенограма черепа.
Рентгенографія виконується в кількох проекціях - передній, задній, бічній та зверху. Передчасно зрощені шви легко ідентифікувати за відсутністю зв'язкових ліній та наявністю кісткових гребенів по лінії шва. Самі шви або не видно, або їхня локалізація показує докази склерозу.
Черепна комп'ютерна томографія з тривимірною проекцією, як правило, не потрібна більшості немовлят. Метод іноді виконується, коли операція розглядається як наступний крок лікування або якщо результати рентгенограми неоднозначні.
Методики корекції патології, можливі ускладнення та наслідки

В останні 30 років у сучасній медицині склалося глибше розуміння патофізіології та лікування краніосиностозу. В даний час хірургія, як правило, залишається основним типом лікування для корекції деформації черепа у дітей зі зрощеннями 1-2 швів, що призводять до потворної форми голови. Для дітей з мікроцефалією, що часто спостерігається при помірному краніосиностозі, хірургія зазвичай не потрібна.
При складанні терапевтичної схеми фахівці обов'язково враховують ряд моментів.
- У пацієнтів з мікроцефалією має бути вивчено причину цього захворювання.
- При першому зверненні вимірюється коло головиу поздовжньому напрямку та надалі проводиться спостереження за змінами. Лікар повинен переконатися у нормальному зростанні мозку у пацієнтів із первинним краніосиностозом.
- Регулярно має проводитися спостереження за ознаками та симптомами підвищеного внутрішньочерепного тиску.
- Якщо є підозра про підвищений внутрішньочерепний тиск, то тут дуже доречно нейрохірургічне консультування.
- Для збереження зорових функцій у пацієнтів із підвищеним внутрішньочерепним тиском слід провести додаткові офтальмологічні консультації.
Хірургічне лікування зазвичай планується при підвищеному внутрішньочерепному тиску або корекції деформації черепа. Операція зазвичай виконується в перший рік життя.
Умови для хірургічного втручання
- Якщо форма голови не змінюється на краще у віці двох місяців, то аномалія навряд чи зміниться з віком. Раннє втручання показано, якщо діти можуть стати кандидатами щодо мінімально інвазивної хірургії. Варто відзначити, що деформація більш помітна у грудному періоді, і вона може стати менш очевидною з віком.
- Коли дитина росте, у неї з'являється більше волосся, видимі прояви аномалії можуть зменшуватись.
- Показання для хірургічної корекції краніосиностозу залежать від віку, загального стану дитини та кількості передчасно зрощених швів.
- Хірургічне лікування черепної чи черепно-лицьової деформації виконується у дітей віком 3-6 місяців, хоча серед хірургів варіюється різноманітність підходів.

Хірургічне втручання у немовлят може призвести до відносно великих втрат крові.Відповідно, слід розглянути мінімально інвазивні хірургічні методи. Одним із перспективних є використання інтраопераційної транексамової кислоти. Пацієнтів із показаннями до хірургічної корекції краніосиностозу піддавали попередньої обробки еритропоетином та транексамовою кислотою, що дозволяло підтримувати нижчі обсяги втрати крові.
Інші особливості оперативного втручання
- Хірургічне лікування у немовлят віком від 8 місяців може бути пов'язане з уповільненням росту черепа.
- Немовлята з діагностованим синдромальним краніосиностозом мають бути прооперовані якнайшвидше.
- Результати операції краще, якщо вона виконується у немовлят віком молодше 6 місяців.
РОЗДІЛ 24 ВРОДЖЕНІ ДЕФЕКТИ ТА АНОМАЛІЇ РОЗВИТКУ ЧЕРЕПА І ГОЛОВНОГО МОЗКУ, ДЗВІНЧНИКА І Спинного МОЗКУ
24.1. ЗАГАЛЬНІ ПОЛОЖЕННЯ
Аномалії(від грец. anomalia - відхилення, мають на увазі відхилення від норми, від загальної закономірності, неправильність) - структурні відхилення від норми, зумовлені порушеннями пренатального розвитку; вони являють собою уроджені дефекти, які виявляються вже при народженні чи ранньому дитячому віці. Виражені аномалії називають пороками розвитку.Пороки розвитку, при яких виявляється спотвореною якась частина організму або все тіло, іноді називають потворністюабо позначають французьким словом «monstre»,проте ці терміни, природно, викликають заперечення з погляду етики та деонтології.
Під вродженими аномаліями маються на увазі відхилення від норми у будові окремих частин тіла, органів та тканин. Можливі вроджені аномалії процесів метаболізму; Наслідком їх можуть бути, зокрема, різні варіанти олігофренії.
За етіологічною ознакою розрізняють 3 групи вроджених аномалій: а) спадкові, що виникли в результаті успадкованих або спонтанних мутацій; спадкові аномалії можна поділити на геномні, хромосомні та генні; б) екзогенні, обумовлені інфекційними або токсичними тератогенними ушкодженнями ембріона або плода та в) мультифакторні. До вроджених аномалій відносяться різні форми порушення розвитку органів та тканин. 1. Агенезія- Повна вроджена відсутність органу. 2. Аплазія- Вроджена відсутність органу за наявності його судинної ніжки.
3. Відсутність або недорозвинення окремих частин тіла та органів, при цьому недостатність їхнього розвитку нерідко позначається складовим терміном, що включає грецьке слово oligos(малий) і назва дефектного органу: наприклад, олігогерія – недостатність мозкових звивин, олігодактилія – недостатня кількість пальців. 3. Вроджена гіпоплазія- недорозвинення органу, що виявляється недостатністю його маси чи розмірів. Розрізняють просту та диспластичну форми гіпоплазії. При простій формі немає якісних змін структури та функцій органу; диспластична ж гіпоплазія позначається на функціональному стані органу (наприклад, диспластична гіпоплазія ока, або мікрофтальм, що супроводжуються розладами зору).
4. Вроджена гіпотрофія- Зменшення маси тіла плода або новонародженого. 5. Вроджена гіперплазія,або гіпертрофія,- Відносне збільшення маси частини тіла або органу. 6. Макросомія (гігантизм)- Збільшення тіла або його частини; зі збільшенням окремих органів чи його частин іноді при-
змінюється грецький термін pachis (товстий): наприклад, пахіакрія - потовщення фаланги пальця, пахігірія - потовщення мозкової звивини. 7. Гетеротопія- наявність клітин, тканин або цілої ділянки органу в іншому органі або в тих частинах того ж органу, в яких їх не повинно бути, наприклад наявність грушоподібних клітин Пуркіньє в зернистому шарі кори мозочка. Гетеротопія тканин характерна для деяких пухлин, наприклад, тератоми, дермоїдні кісти, холестеатоми. 8. Гетероплазія- Порушення диференціювання тканини, також може бути основою пухлинного росту. 9. Ектопія- Зміщення органу, розташування його не на звичайному місці. 10. Подвоєння- Збільшення в 2 рази кількості органів або їх частин; приставка «полі» (від грец. polis - багато) означає збільшення їх кількості в невизначену кількість разів, наприклад полідактилія, полігерія. 11. Атрезія- повна відсутність судини, каналу або отвору, наприклад, атрезія водопроводу мозку, атрезія зовнішнього слухового проходу. 12. Стеноз- звуження судини, каналу чи отвору. 13. Нерозділорганів, частин тіла. Назви аномалій, при яких є нерозділення кінцівок або їх частин, мають приставку "sym" або "syn" (разом), наприклад симподія - нерозподіл ніг, синдактилія - Нерозділення пальців. Можливий і нерозділ двох симетрично або асиметрично розвинених однояйцевих близнюків. Нерозділені двійнята("сіамські близнюки") називають пагами, додаючи до цього слова латинську назву частин тіла, якими вони з'єднані, наприклад, при зрощенні головами - краніопаги (див. рис. 24.3), грудною клітиною - торакопаги і т.п. 14. Персистування- Збереження структур, що в нормі зникають до певного періоду ембріонального розвитку. Персистування ембріональної тканини може стати причиною розвитку пухлин, що виникають внаслідок дизембріогенезу (за теорією Конгейма), наприклад, краніофарингіома. 15. Дизрафія- незарощення ембріональної серединної щілини - незарощення верхньої губи, піднебіння, дужок хребців і т.д. 16. Інверсія- Зворотне (дзеркальне) розташування органів.
Пренатальний, зокрема ембріональний розвиток нервової системи - найскладніший процес, який може бути порушений під впливом різних причин, у тому числі успадкованих особливостей генофонду і ендогенних або екзогенних впливів, насамперед внутрішньоутробних травм, інфекції та інтоксикації. Характер аномалій, що виникають при цьому, багато в чому залежить від фази розвитку нервової системи: стадії формування нервової трубки (перші 3,5-4 тижні), формування мозкових бульбашок (4-5 тижнів), кори великих півкуль (6-8 тижнів) і т.д. Внаслідок цих причин можуть виникати різноманітні дефекти розвитку головного та спинного мозку, черепа та хребта. Ці вади можуть зустрічатися ізольовано або у різних поєднаннях.
Вторинні порушення розвитку та деформації черепа та мозку у внутрішньоутробному періоді, під час пологів або ранньому дитинстві, а також у пізнішому віці можуть бути наслідком травматичних ушкоджень, інфекційних захворювань, а іноді і неуточнених обставин. Вторинні деформації тканин голови та мозку можуть бути зумовлені передчасним зрощенням черепних кісток, гідроцефалією, рахітом, хворобою Педжета, мармуровою хворобою та ін.
Перед порушенням розвитку ЦНС припадає понад 30% всіх аномалій, що виявляються у дітей (Huidi C., Dixian J., 1980). Частота вроджених аномалій розвитку ЦНС варіює, середній показник її 2,16 на 1000 народжених.
24.2. КРАНІОСИНОСТОЗ, КРАНІОСТЕНОЗ
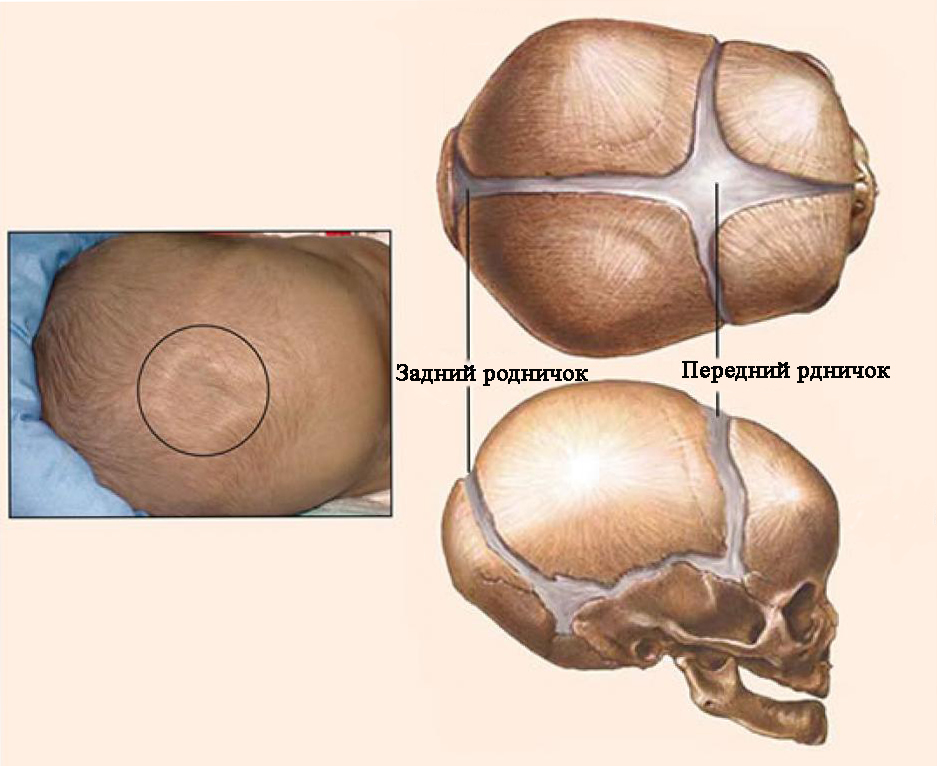
Однією з причин аномалій черепа є передчасне та іноді нерівномірне окостеніння черепних швів.краніосиностоз(Від грец. Kranion – череп і sinostosis – зрощення). У нормі у новонароджених дітей всі кістки склепіння черепа не зрощені, переднє і заднє тім'ячко відкриті. Заднє тім'ячко закривається до кінця 2-го місяця, переднє - протягом 2-го року життя. До кінця 6-го місяця життя кістки склепіння черепа з'єднуються між собою щільною фіброзною мембраною. До кінця 1-го року життя розмір голови дитини становить 90%, а до 6 років вона досягає 95% від розміру голови дорослої людини. Змикання швів шляхом з'єднання зубчастих країв кісток починається до кінця 1-го року життя та повністю закінчується до 12-14-річного віку.
Передчасне та нерівномірне заростання тім'ячків та черепних швів у дітей веде до розвитку краніостенозу(від грец. kranion – череп і stenosis – звуження) і, отже, до недостатності об'єму порожнини мозкового черепа, що перешкоджає нормальному розвитку мозку та веде до створення умов для ліквородинамічних порушень. Частота краніостенозу 1 на 1000 новонароджених. При краніостенозі зазвичай підвищений внутрішньочерепний тиск, у зв'язку з цим характерний гіпертензійний головний біль, можливі розвиток застійних дисків зорових нервів з подальшою їх вторинною атрофією та порушенням зору, відставання у розумовому розвитку (докладніше про внутрішньочерепну гіпертензію див. у розділі 20).
Розрізняють первинний (ідіопатичний) та вторинний краніосиностози. Розвиток вторинного краніосиностозу може бути зумовлений різними причинами. До них можуть бути віднесені вітамін D-дефіцитний рахіт, гіпофосфатемія, передозування тиреоїдного гормону у випадках лікування вродженої гіпотиреоїдної олігофренії (кретинізму).
Заростання швів черепа не тільки передчасне, а й нерівномірне зазвичай веде до черепа деформації. У процесі контролю над розвитком форми мозкового черепа враховується так званий черепний індекс (ЧІ) - співвідношення поперечного розміру черепа до його поздовжнього розміру, помножене на 100. При нормальному (середньому) співвідношенні поперечного і поздовжнього розмірів голови (при мезоцефалії) черепний індекс у чоловіків становить
76-80,9, у жінок – 77-81,9.
При передчасному заростанні сагіттального шва (сагітальний синостоз) розвивається доліхоцефалія,при якій череп збільшується у переднезадньому напрямку і виявляється зменшеним у поперечному розмірі. У таких випадках голова виявляється вузькою та подовженою. ЧІ при цьому менше ніж 75.
Варіант доліхоцефалії, обумовленої передчасним зарощенням сагіттального шва (рис. 24.1), при якому виникає обмеження росту черепа в поперечному напрямку і виявляється надмірним зростанням його в довжину, може бути скафоцефалія(від грец. skaphe - човен), цимбоцефалія(човноподібна голова, кілеголовість), при якій формується довга вузька голова з виступаючими чолом і потилицею, що нагадує перевернутий вгору кілем човен. Сідлоподібнимназивається подовжений у поздовжньому напрямку череп із втиском у тім'яній ділянці.
Варіантом деформації черепа, при якій череп має збільшений поперечний розмір у зв'язку з передчасним зарощенням коронарних швів (коронарний, або вінцевий, синостоз), є брахіцефалія(від грец. brachis - короткий і kephale - голова), голова при цьому широка і
Мал. 24.1.Скафокранія у дитини 5 років.
укорочена, черепний індекс понад 81. При брахіцефалії у зв'язку з двостороннім коронарним синостозом обличчя сплощене, нерідко проявляється екзофтальм.
При передчасному зарощенні вінцевого шва з одного боку розвивається плагіоцефалія,або косоголовість (від грец. plagios - косий і kephale - голова). У таких випадках череп асиметричний, лобова кістка на боці синостозу сплощена, на цій же стороні можливі екзофтальм та збільшення середньої та задньої черепних ямок.
Якщо виникає передчасне поєднане зарощення коронарного та сагітального черепних швів, зростання черепа відбувається в основному в сторони переднього джерельця та основи, що призводить до збільшення висоти голови при обмеженні її зростання у поздовжньому та поперечному напрямках. В результаті формується високий череп конічної форми, дещо сплощений у переднезадньому напрямку. (акрокранія),його часто називають баштовим черепом(Рис. 24.2). Варіант баштового черепа оксицефалія,або гострокінцева голова (від грец. oxys - гострий, kephale - голова), при якій раннє заростання черепних швів веде до формування високого черепа, що звужується догори зі скошеним назад чолом.
Варіант деформації черепа, що характеризується вузькою лобовою та широкою потиличною кістками, формується у зв'язку з передчасним заростанням
лобового шва. Лобові кістки при цьому зростаються під кутом (у нормі заростання лобового шва відбувається лише до кінця 2-го року життя) і на місці лобового шва формується «гребінь». Якщо у таких випадках компенсаторно збільшуються задні відділи черепа та поглиблюється його основа, виникає тригонокранія,або трикутний череп(від грец. Trigonon – трикутник, kephale – голова).
Ізольований синостоз ламбдоподібного шва зустрічається вкрай рідко і супроводжується сплощенням потилиці та компенсаторним розширенням передньої частини черепа зі збільшенням переднього джерельця. Нерідко він поєднується з передчасним закриттям сагіттального шва.

Мал. 24.2.Баштовий череп у дитини 3 років.
Прикладом поєднання генетично обумовленого краніостенозу з іншими патологічними проявами може бути симптомокомплекс Терсиля(описав у 1942 р. французький лікар Thersil M.): баштовий череп, екзофтальм, ністагм, олігофренія, епілепсія, атрофія зорових нервів. На краніограмах зазвичай є прояви внутрішньочерепної гіпертензії, зокрема, виражені пальцеві вдавлення.
При вторинному краніостенозі на ранньому етапі його розвитку може бути ефективним консервативне лікування основного захворювання. При первинному краніостенозі, а також при вторинному краніостенозі в разі значної внутрішньочерепної гіпертензії, що вже розвинулася, показана декомпресивна операція: формування краніоектомічних проходів шириною до 1 см по лінії шовних окостенінь. Своєчасне хірургічне лікування при краніостенозі може забезпечити надалі нормальний розвиток мозку.
24.3. ГІПЕРТЕЛОРИЗМ І ГІПОТЕЛОРИЗМ
Одним із варіантів аномалії черепа є гіпертелоризм(від грец. tele - далеко, horismos - розмежування, поділ), що є наслідком надмірного розвитку малих крил основної кістки. Значно збільшено відстань між внутрішніми краями орбіт, широке перенісся, плоска спинка носа, широко розставлені очі. Може поєднуватися з мікроофтальмією, епікантусом, двостороннім косоокістю, що сходяться, іншими аномаліями, відставанням в психічному розвитку.
Сімейні форми гіпертелоризму успадковуються за аутосомно-домінантним типом. Гіпертелоризм може бути однією з ознак спадкових хвороб, що мають різний тип передачі (синдроми Крузона, Грега, «котячого крику» та ін.).
При гіпертелоризмі індекс міжорбітально-окружний (ІМО) більше 6,8. ІМО дорівнює результату розподілу відстані (у сантиметрах) між внутрішніми кутами очних щілин на коло голови, помноженому на 100.
Гіпотелоризмомприйнято називати зменшення відстані між внутрішніми краями очних ямок; при цьому можливе недорозвинення носа, обличчя схоже на морду мавпи, ІМО менше 3,8. Гіпертелоризм може бути одним із ознак деяких спадкових захворювань, наприклад синдрому Патау.
24.4. МАКРОКРАНІЯ, МІКРОКРАНІЯ, КРАНІОТАБЕС, КРАНІОСКЛЕРОЗ
Збільшення розмірів черепа (макрокранія)може бути не тільки вродженим, а й набутим, наприклад, при рахіті, недосконалості остеогенезу, черепно-ключичному дизостозі.
У новонароджених можлива асиметрична макрокраніята у зв'язку з субдуральною гематомою, гігромою, арахноїдальною кістою в області склепіння черепа. Асиметрія черепа при геміатрофії мозку внаслідок перенесеного в ранньому дитинстві травматичного або запального його ураження, що супроводжується ущільненням, іноді потовщенням кісток склепіння черепа, відоме як
симптом Копилова (Описав вітчизняний нейрорентгенолог Копилов М.Б., нар. 1887 р.). Треба мати на увазі, що асиметрія черепа при народженні може бути наслідком підшкірної або подапоневротичної гематоми.
При рахіті, зазвичай при гострій його течії, іноді виникає краніотабес- розм'якшення, стоншення плоских кісток черепа в області переднього та заднього тім'ячків, над соскоподібними відростками та по ходу черепних швів. Можливий також розвиток гіперостозу черепа (краніосклероз)- повільно прогресуюче потовщення та нерівномірне збільшення розмірів кісток черепа, частіше лицьового; спостерігається, наприклад, при паратиреоїдній остеодистрофі, нейрофіброматозі, еозинофільній аденомі гіпофіза (соматотропіномі), при пухлинах кісток черепа.
24.5. КРАНІОПАГІЯ
Краніопагія відноситься до найбільш рідкісних і небезпечних вроджених каліцтв; вона є зрощення двох однояйцевих близнюків головами (рис. 24.3).
Поділ краніопагів відноситься до найскладніших нейрохірургічних втручань, що включають поділ мозку обох немовлят, кровопостачають їх мозок судин, твердої мозкової оболонки, шкірних покривів і здійснення складних реконструктивних операцій для заміщення неминучих при поділі близнюків дефектів кісток черепа і м'яких тканин. У літературі описано близько 30 операцій із поділу краніопагів, ці операції, на жаль, найчастіше закінчуються загибеллю одного або обох близнюків. Досвід успішної операції з розподілу краніопагів належить Інституту нейрохірургії ім. Н.М. Бурденко РАМН.

Мал. 24.3.Сіамські близнюки, зрощені головами, - краніопаги.
24.6. ПЛАТИБАЗІЯ
Аномалія розвитку черепа, що проявляється сплощенням його основи, - платибазія (від грец. Platys - плоский і basis - основа). Вона може бути і наслідком тривалої внутрішньочерепної гіпертензії, що проявилася в дитячому віці. При платибазії особливо сплощена задня черепна ямка, зазвичай сильно збільшена відстань між спинкою турецького сідла та великим потиличним отвором; кут, утворений схилом черепа (блюменбахів скат) і передньою частиною основи черепа (фронтальна основа, площина передньої черепної ямки), більше 105?; передній край великого потиличного отвору та передня дуга атланта дещо підняті (рис. 24.4б). Платибазія іноді протікає безсимптомно, але може супроводжуватись підвищенням внутрішньочерепного тиску. Вроджена платибазія спостерігається при хворобі Дауна, мукополісахаридозах, може поєднуватися з аномалією Арнольда-Кіарі, ахондропатією. Придбана платибазія можлива при хворобі Педжет, остеомаляції, фіброзної дисплазії, гіпотиреозі, вона може супроводжуватися базилярною імпресією.
24.7. БАЗИЛЯРНА ІМПРЕСІЯ
Базилярна імпресія (базилярна інвагінація, базилярне тиск) зазвичай виникає на тлі вродженої платибазії і є поглибленням переднього відділу основи потиличної кістки (країв великого потиличного отвору, потиличних виростків) у бік субтенторіального простору. На краніограмах при цьому можна відзначити збільшення кута між схилом і верхньою пластинкою основної кістки (більше 130?, рис 24.4в), а також зміщення верхніх шийних хребців, перш за все зуба II шийного (осьового) хребця вище лінії Чемберлена (Умовна лінія, що з'єднує задній край твердого піднебіння з заднім краєм потиличного отвору, що визначається на профільній краніограмі) і лінії де ля Пті (Умовна лінія між верхівками соскоподібних відростків, що визначається на фасній краніограмі). Зазвичай у таких хворих коротка шия, обмеження рухливості, низько розташована межа росту волосся на шиї. У перше-друге десятиліття життя можливі клінічні прояви порушення функцій структур, розташованих у задній черепній ямці, і верхніх шийних сегментів спинного мозку (спастичний тетрапарез, елементи бульбарного синдрому, ністагм при повороті погляду вниз - ністагм, що б'є вниз, та ін.) , а також порушення ліквородинаміки, що проявляються гідроцефалією (див. синдром Арнольда-Кіарі-Соловцева, розділ 11).
24.8. ПІДВИВИВИХ В АТЛАНТООСІВОМУ СУСТАВІ
Чинником ризику є нестабільність в атлантоосьовому суглобі. У таких випадках навіть легка травма може призвести до його підвивиху та глибокого неврологічного дефекту, обумовленого компресією спинномозкових корінців C I -C II та відповідних нервів, а також хребетних артерій та орального відділу спинного мозку. У разі можливого при цьому вклинення

Мал. 24.4.Визначення платибазії та базилярної імпресії.
а - в нормі: тверде піднебіння, верхівка зуба осьового (II шийного) хребця і край великого потиличного отвору розташовані на одній лінії або верхівка зуба осьового хребця знаходиться нижче цієї лінії, а кут, утворений основою передньої черепної ямки та схилом, дорівнює приблизно 105 градусів ; б - платибазія: кут нахилу ската по відношенню до основи передньої черепної ямки понад 105 градусів; в - базилярна імпресія: верхівка зуба осьового хребця вище лінії, що проходить через тверде піднебіння і край потиличного отвору; кут нахилу ската більше 105 градусів.
зубоподібного відростка II шийного (осьового) хребця у великий потиличний отвір зазвичай настає смерть від зупинки дихання. Схильність до підвивиху атлантоосьового суглоба при синдромі Дауна, ревматоїдному артриті, мукополісахаридозі.
24.9. АКРОЦЕФАЛОСИНДАКТИЛІЇ
Багатоваріантну групу вроджених аномалій складають різні форми поєднань баштового черепа (акрокранію, акроцефалію) з різними варіантами аномалії пальців (акроцефалосиндактилії, акроцефалополісіндактилії).
24.10. СИНДРОМ ГРУБЕРА
Серед інших спадкових захворювань, що супроводжуються вираженою кістковою патологією, зокрема змінами черепа, можна відзначити синдром Грубера, що проявляється мікроцефалією, сплощенням очниць, екзофтальмом, вадами розвитку лицьового скелета, нерідко розщепленням дужок хребців, оболонковими та оболонково-мозковими грижами. Цей синдром успадковується за аутосомно-рецесивним типом. Описав його у 1933 р. H. Gruber.
24.11. Закінчені дефекти черепа
На краніограмах іноді вдається виявити невеликі вроджені закінчені дефекти черепа, що локалізуються в сагітальній площині або парасагітально, переважно в тім'яній ділянці. Закінчені дефекти черепа іноді поєднуються з проявами дизрафії, зокрема, дизрафії дуже хребців.
24.12. ДИЗОСТОЗИ ЧЕРЕПА
Деформації черепа можуть бути проявом різних варіантів дизостозу.
Черепно-лицьовий дизостоз Крузона, або «папуга», - краніостеноз, обумовлений поєднанням недорозвинення кісток черепа та передчасним заростанням черепних швів. Виявляється зміною форми мозкового та лицьового черепа, при цьому характерні гіпертелоризм, екзофтальм, страбізм, своєрідна гачкувата форма носа, що нагадує дзьоб орла або папуга. Можливі недорозвинення нижньої щелепи, порушення прикусу: нижні зуби попереду верхніх (прогнання), зниження слуху, пірамідна та мозочкова недостатність, рідше – інші осередкові неврологічні симптоми. Можуть бути різні аномалії кісток тулуба та кінцівок. На очному дні нерідко відзначаються ознаки застою, який може змінити вторинну атрофію дисків зорових нервів, що супроводжується порушенням зору.
Успадковується за аутосомно-домінантним типом. Описав 1912 р. французький лікар O. Crouzon (1874-1938).
Черепно-лицьовий дизостоз Франческетті-Цвалена характеризується грубими порушеннями будови мозкового та лицьового відділів черепа. («Риб'яче обличчя»). Обличчя витягнуте, розріз очей антимонголоїдний, верхня та нижня щелепи з обох боків недорозвинені, відзначаються гіпоплазія структур пірамід скроневих кісток, деформація вушних раковин, виражене зниження слуху, іноді аж до глухоти. Нерідко поєднується з іншими вадами розвитку. Наслідується за аутосомно-домінантним типом.
Черепно-ключично-тазовий дизостоз Шенте-Марі-Сентона - сімейне захворювання, що характеризується запізненням заростання черепних швів і тім'ячків, брахіцефалією, вираженим гіпертелоризмом, гіперостозом дна середньої черепної ямки, відсутністю пневматизації пірамід скроневих кісток, недорозвиненням верхніх щелеп і гайморових пазух, запізнілим (внаслідок чого плечові суглоби можна зближувати на грудях до їх дотику), сколіозом, глибоким поперековим лордозом, іноді розщепленням дужок хребців, спинномозковими грижами. Можливі прояви стискання плечових сплетень. Грудна клітина конічної форми, таз вузький, пізніше окостеніння лобкових кісток, брахідактилія, брахімезофалангія, іноді прогресуюче зниження слуху. При рентгенографії виявляють склероз кісткової тканини, деформації кісток, множинні кісткові шпороподібні потовщення. Успадковується за аутосомно-домінантним типом. Можливі й спорадичні випадки. Описали в 1898 р. J. Shentaner, Р. Marie та R. Sainton.
24.13. ПАТОЛОГІЯ ЧЕРЕПА ПРИ СИСТЕМНИХ
ЗАХВОРЮВАННЯХ КІСТОК
Деякі неврологічні розлади пов'язані з системними захворюваннями кісток, з якими у зв'язку з цим повинен бути знайомий лікар-невропатолог, тому нижче наводиться коротка інформація про таку кісткову патологію.
Для фіброзної остеодисплазії,або хвороби Брайцева-Ліхтенштайна,характерно порушення костеутворюючої функції мезенхіми, що виявляється в одній або кількох кістках, що веде до їх деформації та утворення в них вогнищ розрідження, зазвичай відмежованих від здорової тканини кістки склеротичною облямівкою. Об'єм ураженої кістки при цьому може бути збільшений. Найчастіше уражаються трубчасті кістки, але характерні зміни можуть спостерігатися і в кістках черепа. У таких випадках можливі облітерація придаткових порожнин носа, деформація очних ямок, звуження отворів в основі мозкового черепа та в лицьовому черепі, що веде до порушення функції нервів і судин, що проходять через них. Захворювання, можливо, спадкове, проявляється з дитинства. Описав 1927 р. вітчизняний хірург В.Р. Брайцев (1878-1964), дещо пізніше - американський патологоанатом L. Liechtenstein (1906-1977).
Деформуюча остеодистрофія (хвороба Педжета) частіше проявляється у чоловіків віком 40-60 років, характеризується поступово прогресуючим
потовщенням кіркового шару кісток з розвитком гіперостозів, деформацією, викривленням кісток, безладністю їх структури, утворенням кіст; уражаються кістки мозкового черепа, хребта та довгих трубчастих кісток. Розміри мозкового черепа збільшуються, зовнішня пластинка кісток склепіння черепа подекуди потовщена, гіперостози чергуються з ділянками безладного розрідження кістки. У зв'язку з деформацією кісткових отворів та каналів основи черепа та міжхребцевих отворів порушується функція черепних та спинномозкових нервів, можливі розлади кровообігу. Деформація очних ямок обумовлює екзофтальм. Нерідко спостерігаються ознаки внутрішньочерепної гіпертензії. Хребці сплющені; у трубчастих кістках звужено кістковомозкові канали, можливі патологічні переломи кісток, при цьому лінія перелому чітка, рівна, як при переломі очищеного банана («банановий перелом»); посилено фізіологічні вигини хребта. Процес може бути відносно обмеженим чи поширеним. Вміст кальцію та фосфору в крові нормально або злегка збільшено, активність лужної фосфатази підвищена. Передбачається домінантний тип успадкування із різною експресивністю. Описав хворобу 1877 р. англійський хірург J. Paget (1814-1899).
Мармурова хвороба (хвороба Альберс-Шенберга) - сімейний генералізований остеосклероз, що протікає з лейкемічною реакцією крові у дітей, з анемією та лейкопенією у дорослих, нерідко з атрофією зорових нервів та глухотою. Характерними є деформація мозкового та лицьового черепа, зарощення придаткових порожнин носа щільною безструктурною кістковою тканиною. Через поступове звуження отворів у черепі та міжхребцевих отворів можуть виникати поліморфні прояви ураження периферичної нервової системи як на черепному, так і на рівнях хребта. У хребцях кісткові балки губчастої речовини потовщені та ущільнені. У трубчастих кістках відзначається звуження, а потім і зникнення кістковомозкових порожнин, епіфізи булавовидно потовщені та поперечно вичерчені, є схильність до патологічних переломів. Успадковується за аутосомно-рецесивним типом і тоді, виявляючись у фенотипі в перші роки життя, швидко призводить до смерті, або ж – за аутосомно-домінантним типом, виявляючись у 20-40-річному віці. Описав хворобу 1907 р. H.E. Abers-Schonberg.
Синдром Олбрайту являє собою множинну фіброзну дисплазію кісток, що супроводжується болями та спонтанними переломами; при цьому можливі пошкодження верхньої стінки очної ямки. У разі відзначається односторонній екзофтальм, з тієї ж боці - атрофія зорового нерва, офтальмопарез. Звичайні біль голови, розлад слуху, судоми, олігофренія, гіпертиреоз, зони шкірної гіперпігментації. Виявляється у дитячому віці. У дівчаток при цьому можливе передчасне статеве дозрівання (менструації починаються 5-8 років). Етіологія невідома. Описали синдром у 1937 р. американський ендокринолог F. Albright (нар. 1900 р.) та співавт.
Енцефалоофтальмічна сімейна дисплазія Краузе-Різе - ектомезодермальна дисплазія, що виявляється відразу після народження головним чином неврологічними та офтальмологічними симптомами. Характерні доліхоцефалія, іноді гідроцефалія, потилична або попереково-крижова грижа, мозочкова атаксія, абсанси, олігофренія, дратівливість, а також птоз верхніх повік, страбізм, міопія, відшарування сітківки, катаракта. Можливі розщеплення верхньої губи, твердого піднебіння, вроджені вади серця та інші дефекти розвитку. Успадковується за аутосомно-домінантним типом. Описали
цю форму патології 1946 р. австрійський лікар A.C. Krause та у 1958 р. американський офтальмолог A.B. Reese.
Краніометафізарна дисплазія - дифузне розростання кісткової тканини черепа та метафізів трубчастих кісток. Характерні велика голова, гіпертелоризм, сідлоподібний ніс, широко розставлені зуби. Звуження отворів основи черепа може зумовити ураження черепних нервів та судинні розлади. Ноги зазвичай непропорційно довгі, їх суглобові зони стовщені. Перебіг захворювання повільно прогресує. Успадковується за аутосомно-рецесивним типом. Описав цей патологічний процес у 1957 р. O. Lehman.
Синдром Дзержинського - сімейна гіперпластична періостальна дистрофія, що проявляється комбінацією вад розвитку, при цьому характерні різні варіанти краніосиностозу, базилярна імпресія. Кістки мозкового черепа та обличчя потовщені, ущільнені, ніс різко виступає, потовщені ключиці, грудина, іноді спостерігаються воронкоподібні груди, короткі пальці, їх фаланги потовщені. Синдром, мабуть, спадковий. Описав захворювання у 1913 р. польський лікар В.Е. Дзержинський.
При хронічний ксантоматоз,або хвороби Хенда-Шюллера-Християна,характерна тріада Крісчена: дефекти в кістках черепа, екзофтальм та нецукровий діабет. У черепі, а також у хребцях і трубчастих кістках, розвивається ретикулогістіоцитарна проліферація з утворенням гранульом та подальшим розсмоктуванням кісткової тканини. Над вогнищами кісткової деструкції спочатку виникають щільні болючі вибухання, потім у тій же зоні утворюються кратероподібні заглиблення. Руйнування основи черепа і очних ямок може супроводжуватися опущенням очних яблук. Здавлення гранулематозними масами мозку та черепних нервів веде до розвитку різноманітної неврологічної симптоматики. На краніограмі кістки черепа змінені на кшталт «географічної карти» (у зв'язку з осередками остеопорозу з нерівними контурами). В основі лежить генетично обумовлене порушення ліпоїдного обміну з утворенням пухлиноподібних скупчень жироліпоідних мас у різних органах та тканинах. У крові при цьому виявляються ознаки гіпохромної анемії, підвищено вміст холестерину та ліпопротеїнів. Хвороба проявляється у дитячому віці (до 10 років), частіше у хлопчиків. Успадковується за аутосомно-рецесивним типом. Описав хворобу 1933 р. американський педіатр A. Hand (нар. 1868 р.), потім - американський лікар H.A. Christian (1876-1951) та австрійський рентгенолог A. Schuller (нар. 1874 р.).
Синдром Ван-Бюхема - Спадковий генералізований гіперостоз, що проявляється після настання статевої зрілості помірними ознаками акромегалії. З 3-го десятиліття життя виникають екзофтальм, погіршення слуху, периферичні парези лицевих нервів. На рентгенограмах відзначаються прояви генералізованого гіперостозу, у крові – підвищення рівня лужних фосфатаз, нормальний вміст кальцію та фосфору. Описав синдром 1952 р. голландський терапевт F. van Buchem.
Гіпопластична хондродистрофія є вродженою хворобою, що характеризується порушенням енхондрального остеогенезу. Характерні великий мозковий череп з потилицею, сідлоподібний ніс, прогнатизм, низький зріст (у дорослих до 130 см) в основному за рахунок укорочення кінцівок (мікромієлічний нанізм), короткі кисті, виражений поперековий лордоз. Можливі корінцеві болі, нижній парапарез, обструктивні апное уві сні. При народженні довжина тіла 46-48 см, відзначається значне відставання моторного розвитку, можливе помірне відставання розумово-
го розвитку. На рентгенограмах виявляються диспропорція мозкового та лицьового черепа, сплощення основи черепа, укорочення трубчастих кісток, потовщення клубових кісток, крила яких розгорнуті, звуження хребетного каналу. Тип успадкування аутосомно-домінантний, у 80% випадків захворювання обумовлено новими мутаціями.
Дизрафічний синдром, або синдром Бремера,являє собою комплекс дефектів ембріогенезу, розташованих переважно вздовж середньої лінії: високе піднебіння, розщеплення піднебіння і верхньої губи («вовча паща» та «заяча губа»), нерівномірний ріст і неправильне розташування зубів, деформації черепа, грудної клітки, краніо-вертебральні аномалії, прояви сирингомієлії, деформації хребта, розщеплення дужок хребців (spina bifida), спинномозкові та черепні оболонкові та оболонково-мозкові грижі, додаткові та несиметричні грудні залози, нічне нетримання сечі.
24.14. ЧЕРЕПНО-МОЗКОВІ ГРИЖІ
Вродженою вадою розвитку є черепно-мозкові грижі, які зустрічаються з частотою 1:4000-5000 новонароджених. Ця форма вади розвитку формується на 4-му місяці внутрішньоутробного розвитку. Вона являє собою грижове випинання в ділянці кісткового дефекту, який може бути різним за своїм розміром та формою. Локалізуються грижі зазвичай у місцях з'єднання кісток черепа: між лобовими кістками, біля кореня носа, біля внутрішнього кута ока (передні грижі), в області з'єднання тім'яних кісток та потиличної кістки (Задні грижі). Найчастіше зустрічаються передні черепномозкові грижі (рис. 24.5). По локалізації зовнішнього отвору грижового каналу вони диференціюються на носолобні, носорешітчасті та носоочниць-

Мал. 24.5.Дитина з назоорбітальною грижею та гіпертелоризмом до (а) та після (б) операції.

Мал. 24.6.Дитина з грижею в потиличній ділянці.
ні. Задні черепно-мозкові грижі (рис. 24.6) поділяються на верхні та нижні залежно від того, де розташований дефект у потиличній ділянці: вище або нижче потиличного бугра. Крім названих варіантів черепномозкових гриж, іноді виявляються так звані базальні грижі, при яких є дефект кісток основи черепа на дні передньої або середньої черепних ямок, грижовий мішок випинається в порожнину носа або носоглотки. Рідко зустрічаються черепномозкові грижі в ділянці сагіттального шва.
Основними формами черепно-мозкових гриж є: 1) менінгоцеле,при якій грижовий мішок представлений шкірою та зміненими м'якою та павутинними оболонками, тверда мозкова оболонка зазвичай не бере участі в утворенні грижового випинання, а фіксується до країв дефекту кістки; вміст грижового мішка при цьому є ЦСЖ; 2) менінгоенцефалоцеле- грижовий мішок складають ті ж тканини, а вміст його, крім ЦСЖ, становить тканину мозку; 3) менінгоенцефалоцистоцеле- грижове випинання, в яке, крім тих самих тканин, залучається і частина розширеного шлуночка мозку. З перелічених трьох форм черепно-мозкових гриж частіше зустрічається менінгоенцефалоцеле, нерідко іменується як енцефалоцеле. При гістологічному вивченні грижового мішка і його вмісту виявляються потовщення і ущільнення (фіброз) м'якої і павутинної оболонок, різка атрофія і переродження мозкової тканини, що опинилася в грижовому мішку.
Поверхня грижового випинання може бути покрита незміненою шкірою або витонченою, рубцово-зміненою шкірою, що має синювате забарвлення. Іноді вже при народженні дитини в центрі грижі є лікворний свищ. Нерідко в перші роки життя дитини розміри грижового випинання значно збільшуються, при цьому його шкірні покриви витончуються і покриваються виразками. Можливий і розрив грижового мішка з масивною ліквореєю, небезпечною для життя. До того ж виразки на поверхні грижового мішка та лікворні нориці зачату інфікуються, що може зумовити розвиток гнійного менінгоенцефаліту. Грижове випинання буває на ніжці (завужено в підставі) або має широку основу. В останньому випадку воно нерідко пульсує, а при напруженні дитини - напружується. При пальпації грижове випинання може бути різною щільністю, еластичним, флюктуючим.
Передні черепно-мозкові грижі викликають спотворення обличчя, деформацію очниць, носа, при цьому нерідко відзначаються сплощене широке перенісся, неправильне розташування очних яблук, порушення бінокулярного зору. При назоорбітальних грижах, як правило, виявляються деформація та непрохо-
димість слізно-носового каналу, часто розвиваються кон'юнктивіт, дакріоцистит. Базальні черепно-мозкові грижі, що розташовуються в порожнині носа або носоглотки, на вигляд нагадують поліпи. Якщо грижовий мішок знаходиться в одній половині носа, виникає викривлення носової перегородки; при цьому дихання утруднене, мова невиразна з носовим відтінком.
Дуже великі менінгоенцефалоцеле (є опис передньої черепно-мозкової грижі діаметром 40 см) зазвичай супроводжуються вираженою мозковою патологією, і новонароджені в таких випадках виявляються нежиттєздатними. Доля інших хворих, як правило, залежить від розмірів та вмісту грижового випинання, а також можливості оперативного лікування цієї вади розвитку. Діти нерідко відчувають головний біль, запаморочення. Вогнищева мозкова симптоматика може бути або бути помірно вираженою, проте можливі й вогнищеві неврологічні симптоми, зокрема центральні парези, гіперкінези, розлади координації рухів та ін., ознаки недостатності функцій черепних нервів (I, II, VI, VII, VIII, XII). Можливі епілептичні пароксизми, відставання розумового розвитку.
Черепно-мозкові грижі можуть поєднуватися з іншими вродженими аномаліями: мікроцефалією, краніостенозом, гідроцефалією, мікрофтальмією, епікантусом, вродженим птозом верхньої повіки, аномалією розвитку сітчастої оболонки ока та зорових нервів, колобомами (дефекти тканин) , розщепленням дуже хребців.
Лікування мозкових гриж. Показаннями до невідкладної операції у новонародженого є лікворея з грижового мішка або швидке збільшення розмірів грижі з витонченням її покривів та небезпекою розриву. За відсутності термінових показань до операції дитина має перебувати під наглядом педіатрів, невропатологів, нейрохірургів, які зазвичай спільно вирішують питання можливості надання хворому нейрохірургічної допомоги та визначають найбільш сприятливі терміни операції. Треба мати на увазі, що оперативне лікування черепно-мозкової грижі може бути ефективним і часто призводить до сприятливого результату (рис. 24.5).
Протипоказаннями до операції є запальні процеси в оболонках та головному мозку, виражені неврологічні та психічні розлади (імбецильність, ідіотія), прояви гідроцефалії, тяжкі супутні потворності.
Хірургічне лікування полягає у виділенні та висіченні грижового мішка із збереженням при цьому його вмісту. Важливими етапами операції є герметичне зашивання твердої мозкової оболонки та ретельна пластика кісткового дефекту.
При поєднанні носоочникової грижі та гіпертелоризму виконується складна реконструктивна операція, що включає пластику кісткового дефекту та зближення очних ямок. Потиличні мозкові грижі можуть містити венозні синуси твердої мозкової оболонки, що необхідно мати на увазі при хірургічному втручанні.
24.15. ПОРОКИ РОЗВИТКУ ГОЛОВНОГО МОЗКУ
Пороки розвитку можуть виявлятися у різних поєднаннях. Так, наприклад, при синдром Дуранда-Дзунінаознаки дизрафії поєднуються з гідроцефалією, що супроводжується збільшенням мозкового черепа, агенезією
прозорої перегородки, розщеплення дужок хребців, викривлення стоп і двосторонньої гіпоплазією нирок, що веде до порушення водного обміну. Синдром має сімейний, мабуть, спадковий характер. Описали його у 1955 р. італійські педіатри S. Durand та F. Zunin.
У особливу групу аномалій розвитку можуть бути виділені виражені
вторинні вроджені вади розвитку черепа та мозку, що виникли у різні періоди онтогенезу. Причини таких аномалій різноманітні: захворювання матері в період вагітності, опромінення, травматичні ушкодження плоду, вплив на плід різноманітних токсичних факторів, зокрема алкоголю та численних лікарських препаратів, які мають тератогенну дію. Пороки розвитку ЦНС є наслідком одного або кількох основних патологічних процесів, що порушують розвиток мозку: утворення нервової трубки, поділ її краніального відділу на парні утворення, міграція та диференціація клітинних елементів нервової тканини. Вони можуть виявлятися на трьох рівнях: клітинному, тканинному та органному.
Нижче наводиться опис деяких дефектів розвитку головного мозку та черепа, що виникають у процесі онтогенезу (внаслідок дизембріогенезу).
Аненцефалія- відсутність великого мозку, кісток склепіння черепа і м'яких тканин, що покривають його. На місці мозкової речовини зазвичай розташовується сполучна тканина, багата на кровоносні судини, з кістозними порожнинами, висланими медулярним епітелієм, гліальна тканина, поодинокі нервові клітини, залишки судинних сплетень.
Ексенцефалія- відсутність кісток склепіння черепа (акранія) і м'яких покривів голови, у результаті великі півкулі розташовуються відкрито виходячи з черепа як окремих вузлів, покритих м'якою мозковою оболонкою.
Гідроаненцефалія - повна або майже повна відсутність великих напівкуль при збереженні кісток склепіння черепа і його покривних тканин. Голова при цьому нормальних розмірів або дещо збільшена. Порожнина черепа заповнена переважно ЦСЖ. Довгастий мозок і мозок досить розвинені. Середній мозок та інші відділи головного мозку можуть бути відсутні або представлені рудиментарно. Вперше ця форма пороку була описана Ж. Крювельє в 1835 під назвою «гідроцефалічна аненцефалія».
Поренцефалія істинна - наявність у тканині кінцевого мозку порожнин різних розмірів, вистелених епендимою та сполучених із шлуночковою системою та субарахноїдальним простором.
Поренцефалія хибна - замкнуті порожнини у великому мозку, що не мають епендимної вистилки і є кістами після енцефаломаляції різного походження.
Кістозна дисплазія головного мозку, або поліпоренцефалія, - Природжена дисплазія великих півкуль головного мозку, що характеризується утворенням у ньому множинних порожнин, які зазвичай сполучаються зі шлуночковою системою мозку.
Прозенцефалія- порок розвитку, при якому великі півкулі мозку відокремлює один від одного лише дрібна поздовжня борозна, тому межа між правою і лівою половинками кінцевого мозку нечітка (зустрічається з частотою 1:16 000).
Голопрозенцефалія - порок розвитку мозку, при якому його великі півкулі не розділені і мають вигляд єдиної півсфери, а бічні шлуночки представлені єдиною порожниною. Часто поєднується з іншими вродженими по-
роками. Зазвичай смерть настає невдовзі після народження. Можливо проявом трисомії хромосом 13-15. Пороки кінцевого мозку супроводжуються різними, часом грубими, порушеннями будови обличчя та його кісток, зокрема цебоцефалією, етмоцефалією та циклопією. Діти із циклопією зазвичай народжуються мертвими.
Агірія (лісенцефалія) - недорозвинення звивин великих півкуль, при цьому поверхня їх згладжена (гладкий мозок). При мікроскопії виявляються груба зміна архітектоніки кори великих півкуль, відсутність у ній звичайних клітинних верств. Виявляється вираженим порушенням психомоторного розвитку, поліморфними судомами, парезами чи паралічами. Діти зазвичай помирають протягом першого року життя.
Мікро- та полігірія - порок, при якому на поверхні великих півкуль є безліч безладно розташованих дрібних звивин. Зазвичай мікрогірія проявляється симетрично та супроводжується порушенням пошарової будови кори, що має не більше 4 шарів.
Пахігірія (марогирія) - укрупнення основних звивин, тоді як вторинні та третинні звивини відсутні, борозни при цьому випрямлені, вони короткі та неглибокі. Цитоархітектоніка кори у разі порушена. У білій речовині мозку зустрічаються гетеротопії нервових клітин.
Гіпоплазія, або аплазія (агенезія), мозолистого тіла - часткова або повна відсутність мозолистого тіла. У разі аплазії III шлуночок мозку залишається відкритим. Якщо відсутня лише задня спайка, а саме мозолисте тіло лише вкорочено, це називається гіпоплазією.
Синдром Айкарді- гіпоплазія мозолистого тіла у поєднанні з іншими вадами, зокрема з хоріоретинальними аномаліями, при цьому характерні спазми згинальної мускулатури або міоклонічні напади, множинні лакунарні вогнища в судинній та сітчастій оболонках очей, що виявляються при офтальмоскопії у перипапілярній зоні. Розміри атрофічних хоріоретинальних вогнищ варіюють від невеликих, менше діаметра диска зорового нерва, до діаметра в декілька його діаметрів. Часто є дизрафічні зміни хребта. Можливі розумова відсталість, маятникоподібний ністагм, аномалії розвитку очей (мікрофтальм, колобоми зорового нерва та хоріоїдальної оболонки, ектазія склери та ін.). Описаний синдром тільки у дівчаток, це дозволяє вважати, що хвороба може бути наслідком мутації в Х-хромосомі, яка є летальною при розвитку чоловічого організму. Описав у 1956 р. французький педіатр J. Aicardi.
Мікроцефалія (синдром Джакоміні) - недорозвинення головного мозку, що проявляється при народженні зменшенням його маси та розмірів (рис. 24.7). Мікроцефалія зазвичай поєднується із зменшеним колом голови (не менше ніж на 5 см від середніх показників) та подальшим відставанням зростання мозкового черепа (мікрокранія), при цьому шви його можуть довго залишатися відкритими. Кістки черепа часто потовщені, у них рано формуються диплоїдні канали, внутрішньочерепний тиск не підвищений. При мікрокранії зазвичай відзначається відповідне зменшення розмірів та маси головного мозку – мікроцефалія. Морфологічною її ознакою є недорозвинення та неправильна будова великих півкуль при порівняно нормальній архітектоніці мозочка та стовбура мозку. Дитина з мікроцефалією зазвичай відстає у розумовому, а найчастіше й у фізичному розвитку.
Мікроцефалія може бути первинною (справжньої, генетично обумовленої) та вторинної. Первинна мікроцефалія – наслідок генетичного

Мал. 24.7.Мікроцефалія у дитини 3-х років.
дефекту, що успадковується за аутосомно-рецесивним типом або виникає у зв'язку з хромосомними аномаліями. Вторинна мікроцефалія може бути обумовлена перенесеною внутрішньоутробною інфекцією (краснуха, цитомегаловірусний енцефаліт, токсоплазмоз), інтоксикацією або асфіксією, травмою мозку. При вторинній мікроцефалії у мозку можливі кістозні порожнини, осередки крововиливу та звапніння. Зовнішній вигляд дітей із мікроцефалією своєрідний і характеризується диспропорцією між розмірами мозкового черепа та обличчя. Частота мікроцефалії серед немовлят 1:5000. Серед усіх випадків олігофренії 11% відзначається у хворих із мікроцефалією.
Макроцефалія- Збільшення маси та обсягу головного мозку, а разом з цим і мозкового черепа при народженні, зустрічається значно рідше мікроцефалії. У більшості випадків супроводжується порушенням розташування мозкових звивин, змінами цитоархітектоніки кори, осередками гетеротопії в білій речовині, при цьому зазвичай відзначаються прояви олігофренії, можливий судомний синдром. Причиною макроцефалії може бути поразка паренхіми мозку (ліпоідози). На краніограмах кісткові шви не розширені, шлуночки мозку нормального чи майже нормального розміру. Макроцефалію слід диференціювати від гідроцефалії.
Можлива часткова макроцефалія (Збільшення однієї з великих півкуль), яка зазвичай поєднується з асиметрією мозкового черепа. Гемігіпертрофія черепа за рахунок виривання з одного боку луски скроневої кістки і прилеглих відділів лобової і тім'яної кісток може бути пов'язана з виявленими при краніографії поглибленням і розширенням на цій же стороні середньої черепної ямки, порізністю крил основної кістки. В таких випадках гемігіпертрофія черепа вказує на ймовірність наявності в середній черепній ямці непухлинного об'ємного процесу (гематома, гігрома, ксантома, кістозний арахноїдит і т.п.) і відома як синдром Дайка.
24.16. ПОРОКИ РОЗВИТКУ ШЛУНИКІВ МОЗКУ
Пороки розвитку вентрикулярної системи зазвичай проявляються у сфері її анатомічних звужень. Можливі звуження (стеноз та атрезія)міжшлуночкових отворів, водопроводу мозку (сильвієвого водопроводу), серединної та латеральних апертур IV шлуночка мозку. У таких випадках характерний розвиток внутрішньої гідроцефалії, при цьому у разі атрезії міжшлуночкового
отвори з одного боку виникає асиметрична гідроцефалія. Стеноз або атрезія водопроводу мозку, а також його розщеплення можуть успадковуватися, передаючись аутосомно-рецесивним типом або бути зчеплені з Х-хромосомою. Неповне розкриття апертур IV шлуночка мозку часто поєднується з проявами синдрому Денді-Уокера (Див. 24.18).
Недостатність відтоку ЦСЖ із шлуночкової системи при порушенні прохідності (стінозі) водопроводу мозку та апертур IV шлуночка мозку проявляється, як правило, розвитком внутрішньої рівномірної гідроцефалії,супроводжується розтягуванням, стоншенням та атрофією тканини мозку. Розвитку гідроцефалії нерідко супроводжують деякі аномалії основи черепа і верхнього шийного відділу хребта: платибазія, симптом Кліппеля-Фейля та ін. Можливий також гіперсекреторний або арезорбтивний характер гідроцефалії, обумовленої зазвичай перенесеним запаленням мозкових оболонок. Частота вродженої гідроцефалії 05 на 1000 новонароджених. Докладніше про гідроцефалію див. у розділі 20.
24.17. ФАКОМАТОЗИ
Факоматози (від грец. phakos - пляма, oma - суфікс, що означає "новоутворення", "пухлина", osis - суфікс, що означає "процес", "хвороба") - група спадкових захворювань, при яких спостерігається поєднання уражень нервової системи, шкіри та внутрішніх органів. Характерними проявами факоматозу є ділянки порушеної пігментації покривних тканин (гіперпігментовані або депігментовані плями), крокренові бляшки, фіброми, папіломи, ангіоми, що поєднуються з різноманітними неврологічними, психічними, ендокринними та соматичними порушеннями. Для більшості форм факоматозів характерні затримки розвитку різних функцій, насамперед рухів та інтелекту, а також зниження адаптації до екзогенних та ендогенних факторів, факторів соціального середовища. У тяжких випадках спостерігаються олігофренія, атаксія, епілептичні напади. Описи окремих варіантів факоматозу з'явилися наприкінці XIX ст.
Морфологічною основою факоматозов є (Архіпов Б.А., Карпухіна Л.О., 1996) гамартроми, детерміновані порушеннями росту та диференціювання клітин одного або декількох зародкових листків у ранніх стадіях ембріогенезу. З клітин, які ніби затрималися у своїй диференціації та перебувають у стані «перманентної ембріонізації», утворюються гамартроми, що мають схильність до проліферації та неопластичної трансформації. У зв'язку з цим гамартрому розцінюють як пухлиноподібну вроджену ваду розвитку або ембріональну пухлину з бластоматозними тенденціями (Kousseff B.G. et al., 1990). Гамартроми частіше мають ектодермальне походження і складаються з елементів нервової тканини та шкіри. Звідси й інша назва факоматозів - «Нейроектодермальні дисплазії».Вони можуть поєднуватись з мезодермальними та ентодермальними дисплазіями.
Найчастіше зустрічаються такі ознаки нейроектодермальної дисплазії, як гіпер- і гіпопігментовані плями, плями кольору «кава з молоком», фіброми, папіломи, невуси, нейрофіброми, кортикальні та субепендимальні вузлики ЦНС, факоми, ураження типу «тутової ягоди» на очному дні. Серед мезодермальних дисплазій часто зустрічаються ангіоми, ангіоліпоми, аневризми, ектазії та стенози судин, рабдо- та лейоміоми, дис-
плазії кісткової тканини та ін. Прикладом ентодермальної дисплазії може бути поліпоз різних відділів травного тракту.
У каталозі спадкових захворювань V. McKusik (1967) зареєстровано 54 форми факоматозу. Більшість із них успадковується за аутосомнодомінантним типом.
Нейрофіброматоз, або хвороба Реклінгхаузена, зустрічається найчастіше інших факоматозів (1:4000). У дитячому віці (після 3 років) з'являються множинні бліді, жовто-коричневі (кавового кольору) плями, діаметром від просяного зерна до 15 см і більше, переважно на тулубі та проксимальних відділах кінцівок; нерідко спостерігається генералізована точкова пігментація або ластовиння в пахвових областях. Дещо пізніше з'являються ознаки нейрофіброматозу: множинні щільні пухлини різної величини (частіше діаметром 1-2 см), розташовані по ходу нервових стовбурів (невриноми, нейрофіброми), не зрощені з іншими тканинами.
Пухлини можуть виникати і по ходу черепних нервів (невриноми слухового, трійчастого, язикоглоткового нервів). Нерідко пухлини ростуть із тканини спинномозкових корінців і розташовуються у хребетному каналі, викликаючи здавлення спинного мозку. Пухлини можуть локалізуватися також у очній ділянці, у загрудинному, ретроперитонеальному просторах, у внутрішніх органах, викликаючи відповідну різноманітну симптоматику. Часто розвивається сколіоз, можлива гіпертрофія ділянок шкіри, гіпертрофія внутрішніх органів. В основі захворювання лежать аномалії розвитку екто- та мезодерми. Можлива астроцитарна гамартрома. Успадковується за аутосомно-домінантним типом. Виділяють 2 форминейрофіброматозу: класичну, периферичнуформу (нейрофіброматоз-1),при якій патологічний ген локалізований в хромосомі 17, центральнуформу (нейрофіброматоз-2),патологічний ген розташований у хромосомі 22. Описав хворобу 1882 р. німецький патолог F.D. Recklinghausen (1833-1910).
За матеріалами Інституту нейрохірургії ім. Н.М. Бурденко РАМН при нейрофіброматозі-1,поряд з периферичними невриномами та нейрофібромами, можливі мікроцефалія, пігментовані гамартроми райдужної оболонки (вузлики Лиша), гліоми зорових нервів (Зустрічаються у 5-10% хворих), кісткові аномалії, зокрема дисплазія крил основної кістки, що призводить до дефекту даху орбіти і до пульсуючого екзофтальму, односторонні невриноми слухового (вестибулокохлеарного) нерва, внутрішньочерепні пухлини - менінгіоми, астроцитоми, ома, саркома, лейкемія, клінічні прояви сирингомієлії.
У випадках нейрофіброматозу-2 часто розвивається невринома вестибулокохлеарного черепного нерва, яка при цьому захворюванні часто буває двосторонньою, можливі менінгіома, гліальні пухлини, спинальні невриноми. Можливі також помутніння кришталика, субкапсулярна катаракта.
(Козлов А.В., 2004).
Туберозний склероз (хвороба Бурневілля-Прінгла, синдром Бурневілля-Бресау) - гліоз білої речовини мозку, що проявляється в ранньому дитинстві епілептичними нападами (85%), олігофренією у поєднанні з наростаючою пірамідною та екстрапірамідною симптоматикою, шкірною патологією. У віці 4-6 років на обличчі у формі метелика в області носа зазвичай з'являються множинні жовто-рожеві або коричнево-червоні вузлики діаметром трохи більше 1 мм. аденоми Прінгла, які зазвичай визнаються аденомами
сальних залоз, проте є думка і про те, що вони являють собою гамартрому, що походить з нервових елементів шкіри.
На носі при цьому можливі зміни на кшталт телеангіектазій. Часто зустрічаються ділянки так званою крокреневої шкіри, плями кавового кольору, зони депігментації, поліпи, ділянки фіброзної гіперплазії, можливі гамартроми язика, фіброзні бляшки на шкірі чола, волосистої частини голови та округлі фіброми (пухлини Коена) на пальцях ніг, рідше – рук. Нерідко відзначаються диспластичні риси, вроджені вади розвитку, пухлини сітківки та внутрішніх органів (у серці, нирках, у щитовидній та вилочковій залозах та ін.).
На очному дні можливі драглисті утворення брудно-жовтуватого кольору, що нагадують за формою тутову ягоду, - гліоневроми типу астроцитарної гамартроми, ретинальний факоматоз. Іноді виявляються ознаки застою чи атрофії дисків зорових нервів.
На поверхні мозку спостерігаються поодинокі або множинні гліоматозні вузли, за кольором дещо світліше оточуючого мозку і щільніше його на дотик, можлива їхня кальцифікація. Вузли можуть бути і в білій речовині, підкіркових гангліях, а також у стовбурі мозку та в мозочку.
Трапляються і аномалії розвитку звивин мозку у вигляді мікро-і пахігірії. Захворювання найчастіше носить спорадичний характер. Бляшки досягають діаметра 5-20 мм. У корі великих півкуль і мозочка іноді можуть бути виявлені пластинчасті тільця, що нагадують амілоїд. Відбувається дегенерація клітин кори. При КТ-дослідженні голови нерідко можна виявити кальцифікати та гліальні вузлики в паравентрикулярній ділянці, субепендимарно вздовж зовнішніх стінок бічних шлуночків, у зоні міжшлуночкового отвору Монро, рідше – у мозковій паренхімі. На МРТ головного мозку у 60% виявляються гіпотеденсивні вогнища в одній або обох потиличних частках, що розцінюються як ділянки неправильної мієлінізації (Козлов А.В., 2002).
Визнається, що захворювання успадковується за аутосомно-домінантним типом з неповною пенетрантністю мутантного гена. Описали його 1862 р. французький лікар D.M. Bourneville (1840-1909) та у 1880 р. англійський лікар J.J. Pringle
(1855-1922).
Енцефалотригемінальний ангіоматоз Стерджа-Вебера (шкірно-мозковий ангіоматоз; синдром Штурге (Стерджа)-Вебера; синдром Вебера-Краббе-Осле-
ра- вроджена мальформація мезодермальних (ангіоми) та ектодермальних елементів, що виникла в процесі ембріогенезу під впливом екзогенних та генетично обумовлених причин. Характерна тріада: "полум'яний" невус, епілепсія, глаукома. Вроджена велика судинна пляма (невус) зазвичай локалізована на одній стороні обличчя під час гілок трійчастого нерва. Великі плоскі ангіоми червоного або вишневого кольору на обличчі, що блідіють при натисканні, можуть поширюватися на шкіру волосистої частини голови та шию, зазвичай супроводжуються ангіоматозом мозкових оболонок, частіше в конвекситальній зоні тім'яно-потиличної області, атрофією мозок і вогнищ . Можливі олігофренія, геміпарез, відставання росту паретичних кінцівок, геміанопсія, гідрофтальм. На краніограмах та комп'ютерних томограмах відзначаються осередки звапніння, атрофія мозку, розширення субарахноїдальних просторів.
Захворювання найчастіше носить спорадичний характер. Можливі випадки успадкування як за домінантним, так і за аутосомно-рецесивним типом. На КТ і МРТ зазвичай спостерігаються прояви атрофії речовини мозку, що розширюються.
ня шлуночків мозку та підболочкових просторів. Описали захворювання 1879 р. англійські лікарі W.H. Sturge (1850-1919) та H.D. Weber (1823–1918).
Атаксія-телеангіектазія (хвороба Луї-Бар) характеризується симетричними телеангіектазії, що з'являються у віці 3-6 років, особливо на кон'юнктиві, шкірі обличчя та шиї, що зазвичай поширюються на мозкові оболонки, речовину мозку. Крім того, зазначається підвищена схильність до хронічних запальних захворювань (синусит, пневмонія, бронхоектазії та ін.) у зв'язку з генетично обумовленим порушенням клітинного та гуморального імунітету. За перших спроб дитини до самостійної ходьби виявляються ознаки мозочкової атаксії, яка надалі має наростаючий характер, пізніше з'являються гіперкінези за типом міоклонії або атетозу, сухожильна гіпорефлексія, дизартрія. Можливе ураження черепних нервів, утруднення довільних рухів очей (Окуломоторна апраксія). До 12-15 років виникають порушення глибокої та вібраційної чутливості, наростання атаксії. У пізніх стадіях хвороби у зв'язку з ураженням клітин передніх рогів спинного мозку виникають слабкість та атрофія м'язів, фасцикулярні посмикування. На шкірі з'являються пігментні плями кавового кольору, ділянки гіпопігментації, дерматит себорейний. Поступово розвивається атрофія шкіри, поява сивого волосся відзначається вже у шкільному віці. Характерні затримка психічного та фізичного розвитку, звичайні гіпоплазія мозочка, різкіше виражена в його черв'яку, гіпоплазія вилочкової залози, дисгаммаглобулінемія, ураження ретикулоендотеліальної системи (ретикулези, лімфосаркома та ін.). Прогноз поганий. Причиною смерті найчастіше є хронічні захворювання бронхів та легень, лімфоми, карциноми.
Успадковується за аутосомно-рецесивним типом з високою пенетрантністю мутантного гена. Описала хворобу 1941 р. французька лікарка D. Louis-Bar.
Цереброретиновісцеральний ангіоматоз (гемангіобластоматоз, хвороба Гіппеля-Ліндау) - спадково-сімейний ангіоматоз центральної нервової системи та сітківки очей. Характеризується вродженим недорозвиненням капілярів, компенсаторним розширенням більших судин та утворенням судинних клубочків, ангіом, ангіогліом. Неврологічна симптоматика може бути різноманітною у зв'язку з можливим ураженням великих півкуль, стовбура мозку, мозочка, рідше – спинного мозку.
Характерна тріада: ангіома сітківки, ангіоми головного мозку, полікістоз внутрішніх органів чи ангіоретикулема нирок. На очному дні відзначаються різке розширення і звивистість судин, жовті судинні клубочки в сітківці, пізніше - ексудат і крововилив у сітківці, її відшарування. Часто спостерігаються помутніння склоподібного тіла, глаукоми, іридоцикліту. В результаті згодом настає сліпота. Хвороба Гіппеля-Ліндау зазвичай проявляється у хворих на 18-50 років.
Першими симптомами є ознаки ангіоретикулеми мозочка або сітківки очей. При переважанні клінічних проявів ангіоматозу мозочка хвороба відома як «пухлина Ліндау». Ангіоматоз сітківки зазвичай розглядається як «Пухлина Гіппеля». Можливі ураження внутрішніх органів, що характеризуються аномаліями розвитку та утворенням пухлин: полікістозу нирок, феохромоцитоми, гіпернефроми, кістозних пухлин підшлункової залози, печінки. Успадковується за аутосомно-домінантним типом з неповною пенетрантністю. Описали хворобу 1904 р. німецький офтальмолог Е. Hippel, а 1925 р. шведський патолог А. Lindau (нар. 1898 р.).
24.18. АНОМАЛІЇ ТА ДЕСТРУКЦІЇ НА КРАНІОВЕРТЕБРАЛЬНОМУ РІВНІ
У зоні переходу черепа в хребет часто трапляються краніовертебральні аномалії. Вони можуть зумовити порушення кровообігу в хребетних артеріях, розлад лікворообігу. В результаті прояву різноманітних неврологічних розладів, у тому числі вестибулярні, мозочкові симптоми, ознаки внутрішньочерепної гіпертензії, елементи бульбарного синдрому, зокрема порушення функцій черепних нервів бульбарної групи, корінцеві симптоми на верхньошийному рівні, ознаки пірамідної недостатності, а також порушення чутливості по провідниковому типу. корінцеві симптоми на верхньошийному рівні. Можуть виявлятися різноманітні кісткові аномалії, прояви дизрафічного статусу: базилярне втискання, вистояння верхівки зубоподібного відростка вище ліній Чемберлена і де ля Петі, асиміляція атланта (синдром Ольєнека), феномен проатланту та ін. Для краніоверт е , шийний гіперлордоз; можливі асиметрія обличчя, гіпоплазія нижньої щелепи, готичне піднебіння, розширення хребетного каналу на рівні верхніх шийних хребців, кіфосколіоз хребта, розщеплення дужок хребців, деформація стоп на кшталт «стопи Фрідрейха».
Вроджені аномалії розвитку на краніовертебральному рівні характеризуються дефектами розвитку потиличної кістки та розташованих у задній черепній ямці структур та верхнього відділу хребта та спинного мозку. До них відносяться синдроми Денді-Уокера та синдром Кіарі.
Синдром Денді-Уокера являє собою вроджену ваду розвитку каудального відділу стовбура та черв'яка мозочка, що веде до неповного розкриття серединної (Мажанді) і латеральних (Лушки) апертур IV шлуночка мозку. Виявляється ознаками гідроцефалії, а нерідко і гідромієлії. Остання обставина відповідно до гідродинамічної теорії Гарднера може зумовити розвиток сирингомієлії, сирингобульбії. Синдром Денді-Уокера характеризується проявами функціональної недостатності довгастого мозку та мозочка, симптомами гідроцефалії, внутрішньочерепної гіпертензії. Уточнюється діагноз за допомогою методів, що візуалізують мозкову тканину - КТ- та МРТ-досліджень. Виявляються ознаки гідроцефалії, зокрема виражене розширення IV шлуночка мозку; при МРТ-дослідженні можна виявити деформацію зазначених структур мозку. Описали синдром у 1921 р. американські нейрохірурги W. Dandy (1886-1946) та A. Walker (нар. 1907 р.).
Синдром Кіарі(синдром Арнольда-Кіарі-Соловцева, або синдром церебеломедулярної потворності) - вада розвитку субтенторіальних структур ромбовидного мозку, що виявляється опущенням стовбура мозку і мигдаликів мозочка у великий потиличний отвір. Нерідко поєднується з аномаліями кісток основи черепа та верхніх шийних хребців (платибазією, базилярною імпресією, асиміляцією атланту, синдромом Кліппеля-Фейля), з проявами дизрафічного статусу, зокрема із сирингоміелією, сирингобульбією. При синдромі Кіарі можуть виникати утиски довгастого мозку, структур мозочка, верхніх шийних сегментів спинного мозку, оклюзія лікворних шляхів, що веде до бульбарних, мозочкових та провідникових симптомів, до оклюзійної гідроцефалії. Описали синдром у
1894 німецький патолог J. Arnold (1835-1915) і в 1895 австрійський патолог H. Chiari (1851-1916).
В даний час, ґрунтуючись на результатах МРТ-сканування, деякі автори виділяють два варіанти синдрому Кіарі.
Мальформація I типу (Кіарі I) характеризується зміщенням мигдаликів мозочка до рівня великого потиличного отвору. Можливе опущення довгастого мозку, його подовження та передня компресія довгастого мозку зубоподібним відростком, звуження IV шлуночка мозку та великої потиличної цистерни, ліквородинамічні розлади, ознаки недорозвинення та атипової будови артерій вертебрально-базилярного басейну. У неврологічному статусі можливі окорухові, кохлеарні та вестибуломозочкові, бульбарні, а також провідникові рухові та сегментарні рухові та чутливі порушення. Відсутність неврологічних симптомів, проте можуть проявитися пізніше (іноді на 3-4 десятиліття життя, що свідчить про перехід процесу в мальформацію II типу.
При мальформації II типу (Кіарі II) відзначається протрузія у великий потиличний отвір мигдаликів та черв'яка мозочка, структур довгастого мозку, який набуває S-подібної форми. Характерні спастичний тетрапарез, біль у потиличній ділянці та шиї, мозочкова атаксія, вертикальний «біючий» вниз ністагм, елементи бульбарного синдрому, ознаки сирингомієлії, прояви гідроцефалії, провідникові порушення.
Неврологічна симптоматика при синдромі Арнольда-Кіарі може виявлятися з 5-7 років, іноді пізніше, можливо, у 30-40-річному віці, і має прогредієнтну течію. Прояви аномалії Арнольда-Кіарі нерідко поєднуються з краніовертебральною кістковою аномалією (базилярна імпресія, асиміляція атланту, краніостеноз на кшталт скафокранії та ін.). У діагностиці синдрому Кіарі та визначенні його типу особливо цінною зазвичай є інформація, отримана при МРТ головного мозку та краніовертебральної області, а також при транскраніальній доплерографії (Крупіна Н.Є., 2003).
Симптом Бабчина- атрофія заднього півкільця великого потиличного отвору та внутрішнього гребінця потиличної кістки. Виявляється при краніографії, виконаній у задній напіваксіальній проекції. Описано симптом вітчизняного нейрохірурга І.С. Бабчиним при пухлинах краніовертебральної локалізації.
24.19. ДЕЯКІ ВРОДЖЕНІ ЧИ РАНО ПРОЯВЛЯЮТЬСЯ ФОРМИ УРАЖЕННЯ РУХОВОЇ СФЕРИ
24.19.1. Дитячі церебральні паралічі
Дитячі церебральні паралічі (ДЦП) – гетерогенна група синдромів, які є наслідком ушкоджень мозку, що виникли у внутрішньоутробному, інтранатальному (під час пологів) та ранньому постнатальному періодах. Характерна особливість ДЦП – порушення моторного розвитку дитини, зумовлене насамперед аномальним розподілом м'язового тонусу та порушенням координації рухів (парези, паралічі, атаксія, гіперкінези). Позначені
рухові розлади можуть поєднуватися з нападами епілепсії, затримкою розвитку мови, емоційного та інтелектуального розвитку. Іноді розлади рухів супроводжуються зміною чутливості.
Важливою особливістю ДЦП є відсутність прогресування та можлива, хоч і слабко виражена, тенденція до відновлення наявних ознак патології нервової системи.
Частота ДЦП, за різними даними, становить 25-59 на 1000 новонароджених. За даними Московської дитячої консультативної неврологічної поліклініки, у 1977-1978 роках. вона становила 3,3 на 1000 дитячого населення. Частота ДЦП групи дітей, що народилися з масою тіла менше 1500 р, становить 5-15% (Aziz K. et al., 1994). За даними К.А. Семенової (1994) ДЦП є причиною 24% випадків дитячої неврологічної інвалідності.
Етіологія. Етіологічні чинники різноманітні: захворювання (краснуха, цитомегалія, грип, токсоплазмоз та ін.) та токсикози у матері під час вагітності, аномалії пологової діяльності, акушерські операції та травматичні ураження, крововилив у мозок, асфіксії під час пологового акту, несумісність крові матері та плода, травми та хвороби (менінгіти, енцефаліти) у дитини в ранньому післяпологовому періоді. Можливе поєднання кількох шкідливих факторів.
Причинами вродженого ДЦП можуть бути генетичні детерміновані аномалії формування мозку (дисгенезії мозку), що виникають на різних етапах розвитку. Вони є причиною 10-11% усіх випадків спастичних форм ДЦП. Крім того, причиною ДЦП можуть бути судинно-мозкові порушення у плода або новонародженої дитини, зокрема гіпоксично-ішемічна енцефалопатія, ішемічні та геморагічні інсульти, внутрішньочерепні гематоми.
Патогенез. Патогенні фактори, що діють під час ембригенезу, спричиняють аномалії розвитку мозку. На пізніших етапах внутрішньоутробного розвитку можливе уповільнення процесів мієлінізації нервової системи, порушення диференціації нервових клітин, патологія формування міжнейронних зв'язків та судинної системи мозку. При несумісності крові матері та плода за резус-фактором, системою АВ0 та іншими антигенами еритроцитів в організмі матері виробляються антитіла, що викликають гемоліз еритроцитів плода. Непрямий білірубін, що утворився в процесі гемолізу, чинить токсичну дію на нервову систему, зокрема на структури стріопалідарної системи.
У плодів, що перенесли внутрішньоутробну гіпоксію, до моменту народження захисні та адаптаційні механізми виявляються недостатньо сформованими, істотне значення можуть мати асфіксія та черепно-мозкова травма під час пологів. У патогенезі уражень нервової системи, що розвиваються під час пологів та постнатально, головну роль відіграють гіпоксія плода, ацидоз, гіпоглікемія та інші метаболічні порушення, що ведуть до набряку мозку та вторинних розладів мозкової гемодинаміки та ліквородинаміки. Істотне значення у патогенезі ДЦП надається імунопатологічним процесам: мозкові антигени, що утворюються при деструкції нервової системи під впливом інфекцій, інтоксикацій, механічних пошкоджень мозкової тканини, можуть призвести до появи відповідних антитіл у крові матері, що негативно позначається на розвитку мозку плода.
Патоморфологічна картина. Патоморфологічні зміни нервової системи при ДЦП різноманітні. У 30% дітей є аномалії розвитку
мозку - мікрогірія, пахігірія, гетеротопія, недорозвинення півкуль та ін. , гіпоксією, що виникли в процесі родового акта або токсичним, інфекційно-алергічним, травматичним ураженням мозку у внутрішньоутробному або ранньому постнатальному періодах
Класифікація. Пропонуються різні клінічні класифікації ДЦП. Ми наводимо одну з класифікацій, що здобули широке визнання.
Таблиця 24.1.Синдроми (форми) ДЦП (Miller G., 1998)
Переважними є спастичні форми, решта зустрічається значно рідше.
Клінічні прояви. Виниклий дефект у мозку не тільки негативно позначається на стані новонародженої дитини, а й перешкоджає її нормальному розвитку, насамперед розвитку рухової системи, мови та когнітивних функцій. Клінічна картина у разі може варіювати у великих межах. Важливо пам'ятати, що патологічна постуральна активність, прояви підвищення м'язового тонусу нерідко стають виразними лише до 3-4 місяця життя дитини, інколи ж і пізніше. Для відносно ранньої діагностики ДЦП має значення динамічний нагляд дітей, особливо з неблагополучним акушерським анамнезом, облік у своїй динаміки вроджених безумовних рефлексів, послідовності характеру змін м'язового тонусу становлення реакцій випрямлення і рівноваги.
По переважання тих чи інших неврологічних та психічних функцій Л.О. Бадалян (1984) виділяв такі варіанти ДЦП.
1. Спастична диплегія (синдром Літтля) - Найчастіше зустрічається форма ДЦП. Характеризується тетрапарезом із залученням у процес м'язів обличчя, язика, глотки, при цьому особливо виражені рухові розлади в нижніх кінцівках (прояви нижнього спастичного парапарезу з переважанням напруги м'язів стегон, що приводять, і м'язів-розгиначів гомілки і згиначів стоп. Якщо дитина лежить, ноги у нього При спробі поставити на підлогу ноги у нього перехрещуються, він спирається не на всю стопу, а тільки на передню її частину. Ноги при цьому випрямлені та ротовані всередину. При спробі ходити із сторонньою допомогою дитина здійснює танцюючі рухи, ноги її «перехрещуються», тіло повертається у бік провідної ноги. Нерідко вираженість парезів асиметрична, причому відмінність у можливості активних рухів особливо чітко проявляється у руках.
На тлі диплегії можуть бути хореоатетоїдні гіперкінези, до яких залучаються насамперед мімічні м'язи та м'язи дистальних відділів рук. Діти тяжко переживають наявність рухових порушень, неохоче
вступають у контакт зі здоровими дітьми, краще почуваються в колективі, що складається з дітей із подібними хворобами.
2. Подвійна геміплегія - двостороння геміплегія або, частіше, геміпарез, при якому руки страждають більшою мірою, ніж ноги, або вони уражені приблизно однаково. Можлива асиметрія виразності парезів, при цьому тонус м'язів високий, є поєднання спастики та ригідності зазвичай з переважанням останньої. Реакції рівноваги розвинені замало. Майже завжди виражені елементи псевдобульбарного паралічу, у зв'язку з чим утруднено жування та ковтання, мова. Нерідко відзначаються судомні пароксизми, мікроцефалія. Ця форма ДЦП зазвичай супроводжується найбільш значними проявами олігофренії.
3. Спастична геміплегія характеризується відповідними руховими порушеннями переважно з одного боку. Нерідко рухові розлади більш виражені в руці, вона зігнута у всіх суглобах, кисть у дітей раннього віку стиснута в кулак, у пізнішому віці має форму «руки акушера». Нерідко виникають фокальні епілептичні напади на кшталт Джексона. За допомогою візуалізуючих методів дослідження (КТ, МРТ) в одній із півкуль мозку зазвичай виявляють кісту, рубцеві процеси або прояви поренцефалії. Розвиток інтелекту може бути близьким до нормального.
4. Гіперкінетична форма характеризується переважним ураженням структур стріопалідарної системи. М'язовий тонус мінливий, часто коливається між гіпотонією та нормотонією. На цьому фоні виникають м'язові спазми, що перемежуються, напади підвищення м'язового тонусу по пластичному типу. Активні рухи в таких випадках незручні, супроводжуються зайвими руховими реакціями переважно атетоїдного характеру, при цьому гіперкінези можуть бути переважно в дистальних або проксимальних відділах кінцівок, м'язах шиї, мімічній мускулатурі. Гіперкінези можливі на кшталт атетозу, хореоатетозу, хореї, торсійної дистонії. Часто спостерігаються розлади мови (підкіркова дизартрія). Психічний розвиток страждає менше, ніж за інших форм ДЦП. Ця форма ДЦП зазвичай обумовлена імунною несумісністю крові плода та матері.
5. Мозочкова форма характеризується атаксією, обумовленої головним чином поразкою мозочка та його зв'язків. Може поєднуватись з ністагмом, атонічно-астатичним синдромом, ознаками помірного спастичного парезу у зв'язку із залученням до процесу кірково-підкіркових структур мозку.
Лікування. Лікування, точніше, абілітацію 1 хворого з ДЦП слід розпочинати якомога раніше, при цьому воно має бути комплексним. У ранньому віці мозок дитини пластичний і має значні компенсаторні можливості. Абілітація, розпочата в період формування статичних та локомоторних функцій, дає найбільш суттєві результати. Раннє навчання сенсомоторним навичкам з умовно-рефлекторним їх закріпленням сприяє своєчасному розвитку моторики. Крім того, у ранньому віці спастичні явища виражені ще не різко, відсутні стереотипні патологічні пози, деформації, контрактури, внаслідок чого виробляються легше рухові навички.
1 Абілітація - створення можливостей у розвиток відсутніх раніше видів діяльності.
Важливою частиною комплексного лікування є ортопедичні заходи, профілактика контрактур. Для надання фізіологічного становища окремим частинам тіла широко використовуються лонгети, тутори, шини, валики, коміри та ін. м'язового тонусу, полегшення довільних рухів, розвитку послідовних вікових рухових навичок дитини.
З лікарських засобів у процесі лікування ДЦП застосовуються фармакопрепарати, що покращують метаболічні процеси в мозку, - глутамінова кислота, ліпоцеребрин, церебролізин, препарати з групи ноотропів, вітаміни групи В, ацефен та ін. За показаннями застосовують міорелаксанти, при цьому засобом вибору може бути ботокс ботулінічний токсин). Є позитивний досвід (Бєлоусова О.Д., Темін П.А. та ін., 1999) його введення в двоголовий м'яз плеча, а також у згиначів і розгиначів кисті для зменшення м'язового тонусу та пронаторної установки передпліччя; позитивний ефект дало застосування ботокса тими самими авторами для ліквідації динамічної контрактури в гомілковостопному суглобі. Застосовуються також препарати, дія яких спрямована на пригнічення гіперкінезів, протисудомні засоби, ангіопротектори, антиагреганти та седативні засоби.
Останніми роками розробляються методи соматосенсорної стимуляції. Для цього пропонується зокрема носіння космічного навантажувального костюма «Пінгвін» або його модифікації «Аделі». Застосування навантажувального костюма сприяє виправленню положення центру тяжкості тіла хворого та нормалізації пози стояння. Передбачається (Яворський А.Б. та ін., 1998), що при такому лікуванні може відбуватися перебудова нервових зв'язків у півкулях мозку та зміна при цьому міжпівкульових взаємин.
24.19.2. Спастична сімейна параплегія Штрюмпеля
Хронічна прогресуюча сімейна хвороба докладно описана 1886 р. німецьким лікарем А. Штрюмпеллем (Stumpell A., 1853-1925). В даний час вона розцінюється як група захворювань, що характеризується генетичною гетерогенністю та клінічним поліморфізмом. Хвороба успадковується як за аутосомно-рецесивним, так і за домінантним типом.
Патогенезне вивчений.
Патоморфологічна картина. Відзначається симетрична дегенерація в церебро-спінальних провідних шляхах, що поступово прогресує і розповсюджується знизу вгору. Іноді вона супроводжується дегенеративними змінами в ніжному пучку Голля та спинномозкових трактах. Можливі демієлінізація нервових волокон у ніжках мозку, гліоз та зменшення кількості клітин в екстрапірамідних структурах стовбура.
Клінічні прояви . Зазвичай на другому десятилітті життя з'являються стомлюваність ніг, підвищення в них м'язового тонусу та сухожильних рефлексів. Пізніше з'являються клонуси стоп, патологічні стопні знаки. Згодом ознаки нижнього спастичного парапарезу наростають, при цьому спастичний стан м'язів переважає над м'язовою вираженістю.
слабкості. Протягом багатьох років хворі зберігають здатність до самостійного пересування. Хода у них спастична парапаретична. Через вираженість напруги м'язів стегон хворі іноді при ходьбі перехрещують ноги. У стадії захворювання, що далеко зайшла, можливі захисні рефлекси, ознаки спинномозкового автоматизму, контрактури гомілковостопних суглобів. Елементи спастичності можуть у міру розвитку захворювання виявитися в руках, у м'язах плечового пояса. Можливе зниження вібраційної чутливості на ногах. Інші види чутливості, трофіка тканин та функції тазових органів зазвичай не страждають. Можливі деформація стоп (стопа Фрідрейха), легка мозочкова недостатність, міокардіопатія, зниження когнітивних функцій.
Лікування. Патогенетична терапія не розроблена. Як симптоматичні засоби широко застосовуються міорелаксанти (мідокалм, скутаміл, баклофен та ін.).
24.20. АНОМАЛІЇ ТА ВТОРИННІ ДЕФОРМАЦІЇ ДЗВІНЧНИКА
До краніовертебральних кісткових аномалій відноситься симптом Ольєніка- окципіталізація I шийного хребця (атланта) - зрощення його (асиміляція, конкресценція) із потиличною кісткою. Цей симптом може супроводжуватись ознаками краніовертебральної патології, вертебрально-базилярної судинної недостатністю, порушеннями ліквородинаміки. На спондилограмах іноді виявляється феномен проатланту - Наявність елементів додаткового («потиличного») хребця у вигляді рудиментів передньої дуги, тіла, бічного відділу або задньої дуги. Найчастіше вони перебувають у стані злиття з потиличною кісткою, атлантом, верхівкою зубоподібного відростка II шийного (осьового) хребця, проте можуть зберігатися і у вигляді вільних кісточок, розташованих у зв'язковому апараті між потиличною кісткою та атлантом.
Проявом уродженого кісткового дефекту є аномалія Кімерлі.Борозна хребетної артерії на тильній стороні бічної маси атланта виявляється перетвореною на частково або повністю замкнутий канал у зв'язку з утворенням над нею кісткового містка. Це може зумовити здавлювання хребетної артерії, що проходить через цей канал, і розвиток вертебрально-базилярної судинної недостатності, яка іноді проявляється з юних років. Описав патологію 1930 р. M. Kimerly.
Підвивих та розклинювання атлантоосьового суглоба, або суглоба Крювельє,обумовлені дефектністю його формування та нерідким впровадженням у нього вільних фрагментів проатланту, що веде до розвитку у цьому суглобі ознак деформуючого артрозу. Можливий прояв хвороби Дауна, хвороби Моркіо, ревматоїдного поліартриту, травми шиї. До розвитку підвивиху атлантоосьового суглоба привертають слабкість зв'язкового апарату шиї, гіпоплазія зубоподібного відростка, а також наявність так званої суглобоподібної щілини між зубоподібним відростком та тілом ІІ шийного хребця. Хворі зазвичай відзначають болі в шиї та обмеження рухливості голови, при її поворотах болючість та хрускіт. Неврологічні розлади виникають внаслідок нестабільності в атлантоосьовому суглобі і нерідко провокуються легкою травмою шиї, при цьому можливе зміщення атланту вперед та здавлення верхньошийного відділу спинного мозку.
У випадках ураження двох верхніх шийних хребців при туберкульозній інфекції (хвороба Руста), сифіліс, ревматизм, метастази ракової пухлини на спондилограмах відзначаються відповідні етіологічному фактору зміни у верхніх шийних хребцях, іноді і в потиличній кістці (див. розділ 29).
Хвороба Гризеля (кривошия Гризеля) - Спондилоартрит верхньошийного відділу. Виникає частіше у дітей і натомість інфекційних захворювань, іноді є ускладненням синуситу. Характерно ураження зчленування між атлантом та зубом осьового хребця. Виявляється різким болем і хворобливістю у верхній шийній ділянці, а також протибольової контрактурою м'язів, що прикріплюються до атланту. Характерною є стійка спастична кривошия, при якій голова нахилена у бік вогнища ураження і злегка ротована в протилежному напрямку (див. розділ 29).
Синдром осьового хребця є наслідком аномалії розвитку зубоподібного відростка осьового хребця, служить основою формування синдрому зубоподібного відростка, який виявляється не зрощеним з його тілом та представлений самостійною зубоподібною кісткою (os odontoideum) . Ця кістка вільно зміщується при нахилах голови, звужуючи таким чином хребетний канал, що може зумовити розвиток верхньошийного рівня компресійної мієлопатії; у такому разі можливе виникнення провідникової симптоматики та дихальних порушень, а також поява ознак деформуючого артрозу переважно у бічних атлантоосьових зчленуваннях із збільшенням їх суглобових поверхонь за рахунок кісткових розростань із поступовою міграцією суглобів уперед та вниз, тобто. з формуванням краніовертебрального спондилолістезу. Можуть виникати і прояви судинної вертебрально-базилярної недостатності.
Синдром Кліппеля-Фейля (синдром короткої шиї) являє собою вроджені аномалії та зрощення шийних хребців, що часто поєднується з синдромом Ольєніка. Можлива неповне диференціювання шийних хребців та зменшення їх числа, іноді кількість їх не перевищує чотирьох. У клінічній картині характерна тріада: коротка шия («людина без шиї», «шия жаби»), низька межа зростання волосся на шиї, значне обмеження рухливості голови. У важких випадках підборіддя упирається в грудину, мочки вушних раковин торкаються надпліч, іноді - шкірні складки йдуть від вушних раковин до плечей. Може поєднуватись з гідроцефалією, елементами бульбарного синдрому, вертебрально-базилярною судинною недостатністю, провідниковими порушеннями, високим стоянням лопаток, проявами дизрафічного статусу. За даними рентгенологічних досліджень, виділяють дві крайні форми синдрому Кліппеля-Фейля: 1) атлант злитий з іншими шийними хребцями, загальна кількість яких у зв'язку з цим зменшено, зазвичай їх не більше 4; 2) ознаки синдрому Ольєніка та синостоз шийних хребців, висота їх тіл знижена. Нерідко поєднується з платибазією, можливі інші вади розвитку. Описали синдром у 1912 р. французькі невропатологи М. Klippеl (1858-1942) та А. Feil (нар. 1884 р.).
М'язова вроджена кривошия - укорочення грудино-ключично-соскоподібного м'яза внаслідок осередкового її фіброзу, внаслідок чого голова нахилена в уражену сторону. Причина синдрому заміщення ділянки м'яза, що виникає при цьому, сполучною тканиною невідома.
Конкресценція хребців - зрощення сусідніх хребців у зв'язку з аномалією їх розвитку або внаслідок туберкульозного спондиліту, хвороби Бехтерева, посттравматичного спондильозу та інших патологічних процесів.
Платіспондилія- розширення та зменшення висоти тіл хребців у зв'язку з розвитком у них дегенеративних чи некротичних процесів.
Генералізована платіспондилія (синдром Дрейфуса) - енхондральний дизостоз, що проявляється зазвичай на другому році життя дитини (коли вона починає ходити) болем у спині та слабкістю фіксуючого хребет зв'язкового апарату з подальшим розвитком кіфозу або кіфосколіозу. Характерні короткі шия та тулуб при відносно довгих кінцівках, гіпотрофія та надмірна розтяжність м'язів, розбовтаність суглобів. На спондилограмі видно множинну платиспондилію, при цьому висота тіл хребців може бути зменшена в 2-3 рази, розширення просторів між тілами хребців, можливі зменшені розміри кісток тазу та крижів, уроджений вивих стегна або стегон. Описав синдром 1938 р. французький лікар J.R. Dreyfus.
Остеопатія тіла хребця, що проявляється зазвичай у дітей у 4-9-річному віці, відома як синдром плоского хребця (хвороба Кальве)На спон- дилограмах відзначається остеопороз центральної частини тіла хребця, ущільнення замикальних пластинок з подальшим прогресивним ущільненням (платиспондилія) до 25-30% його вихідної висоти. Сплощений хребець відокремлений від сусідніх розширеними міжхребцевими дисками (див. розділ 29).
Патологічний лордоз та кіфоз хребта. Хребетний стовп у нормі має фізіологічні вигини. Вигин уперед (лордоз) зазвичай має місце на шийному та поперековому рівнях, вигин назад (кіфоз) – на грудному. Надмірна вираженість лордоза веде до збільшення навантаження на задні відділи міжхребцевих дисків, а також на міжхребцеві суглоби, у яких у таких випадках можуть розвиватися дистрофічні явища. При цервікалгії або люмбалгії на відповідному рівні відзначається сплощення лордозу, а іноді і трансформація його на кіфоз. При міопатіях зазвичай посилення вираженості поперекового лордоза.
Патологічний кіфоз характерний для туберкульозного спондиліту, він може виникати при цервікалгії або люмбалгії у хворих на остеохондроз хребта, різко виражений при юнацькому кіфозі, синдромі Ліндемана, синдромі Шейєрмана (Див. розділ 29).
Якщо лордоз та кіфоз можуть бути фізіологічними, то сколіоз- стійкий вигин хребта убік - завжди ознака відхилення від норми. Виділяються 3 ступеня сколіозу: I - виявляється лише при функціональних пробах, зокрема при нахилах тулуба в сагітальній та фронтальній площинах; II - визначається при огляді хворого, що стоїть, зникає при підтягуванні на випрямлених руках, на паралельних брусах або на спинках двох стільців, а також у положенні лежачи на животі; III - стійкий сколіоз, що не зникає при підтягуванні на гімнастичній стінці і т.п. і в положенні лежачи на животі. Рентгенологічно виявляється розширення щілин між тілами хребців на опуклій стороні викривлення хребта при сколіозі нерідко називається ознакою Кона - на ім'я вітчизняного ортопеда І.І. Кона (нар. 1914 р.), який описав цю ознаку як прояв прогресуючого сколіозу. Поєднання кіфозу та сколіозу називається кіфосколіоз.
Синдром ригідного хребта - міопатичний синдром, що поєднується з фіброзом та укороченням аксіальної мускулатури, особливо розгиначів хребта, при цьому порушується згинання голови та тулуба, звичайний сколіоз
грудного відділу хребта з контрактурами проксимальних суглобів кінцівок На ЕМГ відзначаються ознаки ураження клітин передніх рогів спинного мозку та м'язів. Характерні м'язова слабкість, міогіпотрофія, ознаки кардіоміопатії та зміна активності креатинфосфокінази. Успадковується за аутосомно-рецесивним або Х-зчепленим рецесивним типом. Описали 1865 р. англійський лікар V. Dubowitz, а під назвою «вроджений артрогрипоз хребта» 1972 р. - вітчизняна невропатолог Ф.Є. Горбачова.
Деформації хребта можуть виникати у процесі інволюції. Такі зміни форми хребта спостерігаються, зокрема, при синдромі Форестьє,що виявляється у осіб 60-80-річного віку, при цьому характерна кругла стареча спина.
При надмірному поперековому лордозі внаслідок тиску остистих відростків одна на одну можлива їхня деформація (Синдром Бострупа, «цілується» остистий відросток).Виявляється болем у ділянці нирок при розгинанні хребта. На спондилограмах виявляються несправжні суглоби між сплощеними остистими відростками.
Сплощення тіла хребця та загострення при цьому його передньої частини є одним з проявів остеохондродистрофії, відоме як деформація Моркіо-Брейлсфорд.
Про останні три клінічні феномени див. також у розділі 29.
24.21. ДИЗРАФІЇ ХРЕБЛЯ І Спинного мозку, спіномозкові брижі
Спінальна дизрафія є пороком розвитку, пов'язаним з неповним закриттям тканин мезодермального і ектодермального походження вздовж серединного шва (від грец. rhaphe - шов) - середньої лінії хребта. Проявами спинальної дизрафії є розщеплення дуг хребців (spina bifida) і сагіттально розташованих м'яких тканин, а також різні варіанти спинномозкових гриж, що виникають при цьому, іноді дермоїдні кісти, ліпоми, синдром «жорсткої» кінцевої нитки.
Дизрафія хребта та спинного мозку залежно від ступеня їхнього недорозвинення має такі варіанти: 1) spina bifida occulta; 2) spina bifida complicata; 3) spina bifida anterior; 4) спинномозкові грижі: менінгоцеле, менінгорадикулоцеле, мієломенінгоцеле, мієлоцистоцеле; 5) рахісхізіс частковий та повний.
Прихована ущелина хребта - spina bifida occulta (Від лат. spina- ость, bifidus - надвоє розділений). Найбільш часта форма аномалії хребта - розщеплення дуг хребців (Spina bifida occulta). Незарощені можуть бути 1-2 хребці, але іноді і більша їх кількість. Кінці незарощених дуг нерідко вдавлюються в просвіт хребетного каналу і викликають компресію твердої мозкової оболонки, субдурального простору та корінців кінського хвоста, при цьому кістковий дефект прикритий незміненими м'якими тканинами. Така форма аномалії виявляється при спондилографії, частіше на нижньопоперековому – верхньохрестцевому рівнях. У зоні розщеплення дуги або кількох дуг хребців іноді відзначається втягнутість і атрофія шкіри або припухлість тканин, рубці, пігментація, можливий гіпертрихоз.
симптом Фавна. Наявність spina bifida occulta може привертати до розвитку больового синдрому, іноді - синдрому Лермітта, що супроводжується відчуттям за типом проходження електричного струму вздовж хребта при постукуванні по остистому відростку аномального або пошкодженого хребця.
Повний рахісхізис - виражена дизрафія, що проявляється розщепленням як дуг і тіл хребців, а й прилеглих до них м'яких тканин. Через ущелину в м'яких тканинах відразу після народження дитини може бути видно спинний мозок. Грижового випинання тканин при цьому немає. Тіла хребців у вентральній частині ущелини можуть зростатися. Можливі вади розвитку та інших хребців, ребер. Зустрічаються парціальні, субтотальні та тотальні форми дизрафії.
Spina bifida anterior- незарощення тіл хребців. Зустрічається рідко і переважно є випадковою знахідкою на спондилограмах, проте може поєднуватися з іншими дефектами розвитку.
Spina bifida complicata- незарощення дуг хребця в поєднанні з пухлинно-видними розростаннями, що являють собою лише жирову або фіброзну тканину, розташовану під шкірою і заповнює кісткові дефекти дуг хребців, зростаючись при цьому з мозковими оболонками, корінцями і спинним мозком. Локалізується частіше на попереково-крижовому рівні хребетного стовпа.
Спинномозкові грижі, що виникають у зв'язку з незарощення дуг хребців і розщепленням м'яких тканин, являють собою вроджені грижові випинання вмісту хребетного каналу (рис. 24.8): менінгоцеле - грижове випинання з мозкових оболонок, заповнене ЦСЖ; менінгорадикулоцеле - грижа, що складається з мозкових оболонок, спинальних корінців та ЦСЖ; мієлорадикуломенінгоцеле - грижа, що включає структури спинного мозку, спинномозкові коріння, мозкові оболонки та ЦСЖ; мієлоцистоцеле - грижовий мішок, що містить ділянку спинного мозку з ознаками гідромієлії.
Діагностика. При спинномозкових грижах діагностика не становить труднощів. Про характер вмісту грижового мішка можна судити на ос-

Мал. 24.8.Дитина зі спинномозковою грижею (мієломенінгоцеле) та супутньою гідроцефалією.
новленні вивчення неврологічного статусу. Уточнення діагнозу може бути забезпечене за допомогою спондилографії та МРТ-дослідження, при цьому треба мати на увазі, що окостеніння крижів відбувається лише приблизно до 12 років життя.
Лікування спинномозкових гриж. Можливе лише хірургічне лікування. При швидкому збільшенні, витончення і виразки покривних тканин грижового випинання, що загрожують його розривом, а також наявністю лікворного свища, показана термінова операція. В іншому випадку можливий розвиток менінгіту, менінгомієліту, менінгомієлоенцефаліту. Протипоказанням до операції можливо запалення тканин хребетного каналу, виражені неврологічні розлади. Питання операції має вирішуватися спільно педіатром, невропатологом і нейрохірургом.
Грижове випинання виділяється з м'яких тканин, стінка його розкривається. Якщо в порожнину грижі випинаються коріння і тканина спинного мозку, то їх по можливості з дотриманням максимальної обережності виділяють зі зрощень і переміщують у просвіт хребетного каналу. Після цього грижове випинання січуть і послідовно пошарово зашивають м'які тканини. При великих дефектах іноді роблять переміщення м'язів та апоневрозу з прилеглих областей для повноцінного закриття дефекту та запобігання повторним випинанням. Якщо грижовий мішок потрапляє спинний мозок, зазвичай, можлива лише паліативна операція.
При лікуванні спинномозкових гриж слід зважати на той факт, що вони нерідко поєднуються з гідроцефалією. У цих випадках, крім видалення грижового випинання, показана операція, що шунтує, - люмбоперитонеостомія.
24.22. АНОМАЛІЇ Спинного мозку
Діастематомієлія - поділ спинного мозку по довжині на дві частини кісткової, хрящової або фіброзної перемичкою. Для такої форми аномалії немає облігатних ознак, оскільки наявні симптоми можливі і за інших вад розвитку хребта і спинного мозку. Проте діастематомієлія може супроводжуватися шкірними проявами, відхиленням від норми у стані опорно-рухового апарату та неврологічними розладами.
При зовнішньому огляді пацієнта з діастематоміелією вздовж осі хребта в зоні розщеплення спинного мозку можуть бути видні ділянки гіпертрихозу, пігментні плями кавового або темно-коричневого кольору, ангіоми, а також втягнуті ділянки рубцово-зміненої шкіри.
Зміни опорно-рухового апарату можливі вже у ранньому дитинстві. Можливі, зокрема, деформації стоп. Слабкість однієї або обох ніг, асиметрія м'язів нижніх кінцівок, гіпотрофія окремих м'язів або м'язових груп, слабкість м'язів ніг та тазового пояса. Нерідко в дітей віком із раннього віку виявляються сколіоз та інші форми деформації позвоночника.
З неврологічних симптомів можуть спостерігатися асиметрія або відсутність сухожильних рефлексів, частіше п'яткового (ахіллова) або колінного, зниження чутливості, ознаки порушення вегетативної іннервації.
Іноді значні за ступенем вираженості ознаки нижнього парапарезу поєднуються з розладом функцій тазових органів, у своїй можуть бути імперативні позиви на сечовипускання, нічне нетримання сечі.
Амієлія- повна відсутність спинного мозку, при цьому збережені тверда мозкова оболонка та спинномозкові ганглії. На місці спинного мозку можливий тонкий фіброзний тяж.
Дипломієлія- подвоєння спинного мозку лише на рівні шийного чи поперекового потовщення, рідше - подвоєння всього спинного мозку.
262. У новонародженої дитини відкриті шви:
1) стрілоподібний *
3) вінцевий *
4) потиличний *
263. Закриття швів черепа у доношених дітей відбувається до:
264. Максимальні терміни закриття великого джерельця припадають на вік:
3) 12-18 міс *
265. Вік закриття малого джерельця:
1) 6 місяців
2) 1-2 місяці
3) 12-15 місяців
4) до народження
5) 12-18 місяців
6) 4-8 тижнів *
266. Вік закриття великого тім'ячка в міс.:
4) до народження
267. Відповідність кількості молочних зубів віку дитини розраховується за формулою (n - вік дитини на міс.):
268. Прорізування всіх молочних зубів закінчується віком:
1) 1-1,5 роки
2) 1,5-2 роки
3) 2-2,5 роки *
4) 2,5-3 роки
269. Перші постійні зуби з'являються у віці:
270. Порушення другої стадії остеогенезу (мінералізація кістки) виникає при:
1) зниження м'язового тонусу *
2) зрушенні рН у кислу сторону *
3) зсув рН в лужний бік
4) дефіцит кальцію, фосфору та інших мінералів *
5) гіповітаміноз Д *
6) гіпервітаміноз Д
271. Перемоделювання та самооновлення кістки:
1) регулюється паратгормоном *
2) регулюється тироксином
3) регулюється тиреокальцитоніном *
4) залежить від забезпеченості вітаміном Д*
5) не залежить від забезпеченості вітаміном Д
272. При гіпокальціємії розвиваються такі механізми:
1) вимивання кальцію з кісток*
2) посилення кишкового всмоктування кальцію *
3) зменшення кишкового всмоктування кальцію
4) посилення ниркової екскреції кальцію
5) зменшення ниркової екскреції кальцію *
273. Найменша ламкість кісток у дітей раннього віку обумовлена:
1) великим вмістом щільних речовин
2) меншим вмістом щільних речовин *
3) великим вмістом води *
4) волокнистою будовою кістки *
5) більшою податливістю при стисканні *
274. Визначення кісткового віку має значення для:
1) оцінки біологічного віку *
2) оцінки нервово-психічного розвитку
3) оцінки відхилення в зростанні та розвитку *
4) оцінки статевого розвитку
5) оцінки відхилення в масі
6) контролю за застосуванням стероїдів *
275. У новонародженої дитини стан м'язів характеризується:
1) м'язової гіпотонії
2) переважанням тонусу м'язів-розгиначів кінцівок
3) переважанням тонусу м'язів-згиначів кінцівок *
4) під час сну м'язи розслаблюються *
5) основна маса м'язів посідає м'язи тулуба *
276. У дітей вміст загального кальцію в сироватці крові становить ммоль/л:
277. З боку грудної клітки при рахіті спостерігаються:
1) рахітичні чотки *
2) Гарісонова борозна (перипневмонічна борозна) *
3) грудна клітина має вигляд "курячої" *
4) лійкоподібна грудна клітка
5) збільшення кривизни спини *


